

Abstract
Purpose:
Structural variants (SV) of the MYC gene region are common in multiple myeloma and influence disease progression. However, the prognostic significance of different MYC SVs in multiple myeloma has not been clearly established.
Experimental Design:
We conducted a retrospective study of multiple myeloma comparing MYC SV subtypes identified by next-generation sequencing (NGS) and FISH to MYC expression and disease survival using 140 cases from Mayo Clinic and 658 cases from the MMRF CoMMpass study.
Results:
MYC SVs were found in 41% of cases and were classified into nine subtypes. A correlation between the presence of a MYC SV and increased MYC expression was identified. Among the nine MYC subtypes, the non-immunoglobulin (non-Ig) insertion subtype was independently associated with improved outcomes, while the Ig insertion subtype, specifically involving the IgL gene partner, was independently associated with poorer outcomes compared with other MYC SV subtypes. Although the FISH methodology failed to detect approximately 70% of all MYC SVs, those detected by FISH were associated with elevated MYC gene expression and poor outcomes suggesting a different pathogenic role for FISH-detected MYC subtypes compared with other MYC subtypes.
Conclusions:
Understanding the impact of different MYC SVs on disease outcome is necessary for the reliable interpretation of MYC SVs in multiple myeloma. NGS approaches should be considered as a replacement technique for a more comprehensive evaluation of the multiple myeloma clone.
Translational Relevance.
Although structural variants (SV) involving MYC are common in multiple myeloma, the prognostic significance of different MYC SVs has not been clearly established. Here, we evaluated the significance of MYC SVs in multiple myeloma detected by next-generation sequencing (NGS) and FISH. MYC SVs were common and nine different subtypes could be identified by NGS. Only non-immunoglobulin (non-Ig) insertion and Ig insertion subtypes showed significant differences in outcome. Non-Ig insertion was associated with improved outcome, while the Ig insertion subtype (specifically IgL) was associated with reduced outcome. Although FISH is commonly used to detect MYC SVs, FISH failed to identify approximately 70% of MYC SVs. MYC SVs detected by FISH were more likely to be associated with higher MYC gene expression and poor outcome. Genome-wide NGS approaches should be considered as a replacement technique for a more comprehensive evaluation of the tumor clone, in comparison with traditional cytogenetic methodologies such as FISH.
Introduction
Multiple myeloma is an incurable malignancy of plasma cells with approximately 32,000 new cases in the United States each year (1, 2). Genetic abnormalities of multiple myeloma contribute to disease heterogeneity and influence response to therapy and prognosis (1). Primary genetic abnormalities, characterized by recurrent immunoglobulin (Ig) heavy chain (IgH) structural variations (SV) and/or hyperdiploidy resulting from multiple trisomies, occur early in disease course, while secondary genetic abnormalities such as 1q gain, 17p deletion resulting in loss of TP53, and SVs involving the MYC proto-oncogene occur upon disease progression (3). Genetic events t(11;14)(q13;q32) CCND1/IgH, t(6;14)(p21;q32) CCND3/IgH and hyperdiplody are associated with standard risk, whereas t(4;14)(p16.3;q32) FGFR3/MMSET/IgH, t(14;16)(q32;q23) IgH/MAF, t(14;20)(q32;q12) IgH/MAFB, 1q gain and TP53 deletions and single-nucleotide variants (SNV) are associated with high risk (3).
Although genetic abnormalities involving MYC have been documented in the progression from smoldering multiple myeloma (SMM) to multiple myeloma (4–6), the prognostic role of MYC SVs has not been fully established in the context of multiple myeloma. Earlier studies showed MYC SVs detected by FISH or target capture–based sequencing were independently associated with poor outcome (7–10). However, more recent studies using whole-genome sequencing (WGS) did not uniformly support this finding (excluding poor outcome MYC/IgL SVs; refs. 4, 11–13). This discrepancy is likely due to differences in methods and sensitivities of detection of MYC SVs by FISH or target capture–based sequencing compared with WGS. Given that MYC SVs often display remarkable genomic heterogeneity with numerous gene partners, reduced detection of MYC SVs by FISH is not unexpected (4, 8, 9, 11, 12, 14). This possibility is supported by lower frequencies of MYC SVs found by FISH (∼13%–15%) compared with next-generation sequencing (NGS; ∼23%–42%) consistent with a high false-negative rate of the MYC break apart (BAP) FISH probe (15). To more fully understand the role of MYC SVs in multiple myeloma disease outcome, we performed a retrospective study to compare the MYC SV subtypes identified by FISH and NGS to the expression of MYC and overall disease survival.
Materials and Methods
This was an institutional review board (IRB)-approved retrospective study that included newly diagnosed multiple myeloma (NDMM) cases from both the Mayo Clinic and publicly available Multiple Myeloma Research Foundation (MMRF) CoMMpass cases.
Mayo clinic cohort
The Mayo Clinic (Rochester, MN campus) cohort included 1,342 unique patients with multiple myeloma seen within 90 days from diagnosis in the period from January 2006 to January 2018. Cytogenetic analysis by FISH, including the MYC BAP, was performed within 1 year from diagnosis and less than 6 months from the start of first-line treatment. All patients were identified using a prospectively maintained database; additional clinical and laboratory data were obtained by review of electronic medical records. All patients had authorized the use of their electronic medical record data for research. Patient samples were collected with written informed consent in accordance with recognized ethical guidelines and IRB approval. Among those patients, 140 had simultaneous data for both FISH and NGS using mate pair sequencing (MPseq).
MMRF CoMMpass cohort
The MMRF CoMMpass cohort (clinical trial identifier: NCT01454297) included 658 cases with either tumor long-insert whole-exome sequencing (WES), WGS, RNA sequencing (RNAseq) for gene expression (IA15 release), and clinical outcome data (IA16 release). The study was approved by ethics committees or IRBs at the study sites. All patients provided written informed consent in accordance with recognized ethical guidelines. RNAseq data from the CoMMpass cohort are displayed as Salmon TPM and log2-transformed [log2(MYC TPM + 0.25)−log2(0.25)] values.
FISH
FISH analysis was performed on bone marrow samples as described previously (16,17) and further described in Supplementary Methods.
DNA extraction and MPseq
DNA extraction and MPseq library preparation methods have been described previously (18–20) and further described in Supplementary Methods.
Bioinformatics and visualization
A total of 658 CoMMpass cases and 200 normal peripheral blood samples with WGS data were analyzed for breakpoint junction (SV) and copy-number abnormality (CNA). The sequencing FASTQ files were aligned to GRCh38 reference genome using BWA-MEM 0.7.17 (21). The output BAM was processed to determine coverage (reads/Kb) across the genome and normalized using a GC content and mapability (Umap k24; ref. 22) correction and then binned to 30 Kb windows. Regions of similar copy-number level were segmented using a sliding window method and then the copy-number value of each region was normalized by the mode of the coverage probability distribution function to center the values around the expected 2N level. CNA regions were calculated as any region that deviated from this expected 2N level by >10% (loss ≤ −10% deviation, gain ≥ +10% deviation). CNA of TP53 and CKS1B were identified as any region that deviated from this expected 2N level by ≤1.74 as loss and ≥2.3 as gain to call TP53 deletions and 1q gains. For SV detection, reads that mapped to locations ≥5 Kb bp apart or to different chromosomes were considered discordant. These discordant fragments were clustered by both fragment size (absolute difference in genomic positions) and midpoint (sum of genomic position). Clustering was done on both parameters using a cutoff of 5 Kb. The clusters from the normal peripheral blood WGS samples were used to create a 5 Kb mask to eliminate likely false positive from the clustering results. A further filtering was applied that required junction calls to have ≥3 supporting fragments to be called and have a genomic footprint of ≥50 bp on both sides of the junction. These SV and CNA calls were combined and visualized in a genome U-plot (19), which allowed us to interpret and characterize the MYC alterations into distinct structural motifs. MYC SV partner frequencies were calculated with R (https://www.R-project.org) and plotted using the ggplot2 library (23) or Circos (24).
ROC
The ROC curve (25) was created by plotting the false-positive rates versus true-positive rate using RNA expression level (transcription per million; TPM) and MYC alterations were classified using WGS data from the 658 samples from the CoMMpass cohort.
Statistical analysis
All statistical analyses were performed using statistical software JMP (Version 14.1.0, SAS Institute Inc.), SPSS15, or GraphPad Prism software and significance was determined at P < 0.05. Categorical variables were compared using the χ2 test. A nonparametric (Kruskal–Wallis test) and post hoc (Dunn) test was used to determine difference between more than two sample groups. Overall survival (OS) was defined as time from diagnosis to death from any cause or to last follow-up, with those alive censored at date of last follow-up. Progression-free survival (PFS) was defined as the time from initiation of first-line treatment until progression or death from any cause, or last follow-up (Mayo Cohort) or from time of diagnosis until progression or death from any cause, or last follow-up (CoMMpass cohort). Survival curves were estimated using Kaplan–Meier and compared using the log-rank test. Where survival probability did not reach 0.5, mean survival times were calculated.
Data availability statement
The MPseq data that support this study have been deposited in the National Center for Biotechnology Information (NCBI)'s Sequence Read Archive with BioProject ID PRJNA739382 and can be accessed at http://www.ncbi.nlm.nih.gov/bioproject/739382. All CoMMpass data used in this study are publicly available [database of Genotypes and Phenotypes (dbGap): phs000748.v1.p1 and EGAS00001001178] and also http://www.ncbi.nlm.nih.gov/bioproject/248538.
Results
Characterization of MYC SV subtypes
A total of 658 cases from the CoMMpass cohort with both WGS and MYC expression data were included, and the associated abnormality impacting the MYC gene region was investigated. Forty-two cases (6.4%) had a CNA involving MYC without a MYC SV, including 18 cases (2.7%) with monosomy 8 (one copy of the MYC gene) and 24 cases (3.6%) with trisomy 8 (three copies of the MYC gene; Table 1). Nineteen cases (2.9%) with a deleterious SNV impacting the MAX gene were included in a separate category given the extremely low MYC gene expression as previously reported (Table 1; ref. 4). We found 327 cases (49.7%) had neither a MYC SV, CNA, nor deleterious MAX SNV and 270 cases (41.0%) had evidence of an SV impacting the MYC gene region. We further categorized the cases with a MYC SV into nine subtypes based on the type of MYC SV observed (Table 1; Supplementary Fig. S1). These included cases with terminal tandem duplications (TTD; duplication of the genomic segment downstream of MYC; n = 29, 4.4%), terminal deletions (terminal del; deletion of the genomic segment downstream of MYC; n = 11, 1.7%), proximal deletions (proximal del; deletion of the genomic segment upstream of MYC; n = 13, 2.0%), translocations involving immunoglobulin (Ig; IgH, IgK, or IgL) enhancer sequences (Ig translocation; n = 13, 2.0%), templated insertions of non-Ig enhancers (non-Ig insertion; n = 65, 9.9%), translocations involving non-Ig enhancer sequences (non-Ig translocations; n = 16, 2.4%), complex deletions or gains (complex del/gain; n = 30, 4.6%), templated insertions of Ig (IgH, IgK, or IgL) enhancer sequences (Ig insertion; n = 88, 13.4%), and amplifications of MYC (n = 5, 0.8%; Table 1). Of 270 cases with a MYC SV, 37.4% involved an Ig enhancer and 62.6% involved a non-Ig enhancer. From 658 cases, 356 (54.1%) were hyperdiploid without evidence of a translocation to CCND1, CCND3, MAF, MAFB, or FGFR3/MMSET. Similar to other studies (4, 7, 9, 11, 12, 14, 26), an increase in the prevalence of hyperdiploidy in cases with a MYC SV was observed (n = 191, 70.7%), compared with cases without a MYC abnormality (n = 165, 42.5%). Specifically, cases with TTD, non-Ig insertion, and Ig insertion had an enrichment of hyperdiploidy (Table 1).
Table 1.
MYC subtype and expression.
| MYC subtype | Median raw TPM | Median log-transformed TPM | Total (frequency in cohort) P < 0.0001 | Total hyperdiploidy (frequency in subtype) | |||
|---|---|---|---|---|---|---|---|
| No MYC abnormality | No MYC SV | 21.6 | 6.4 | 327 (49.7%) | 388 (59.0%) | 142 (43.4%) | 165 (42.5%) |
| MAX SNV | 1.1 | 2.4 | 19 (2.9%) | 2 (10.5%) | |||
| Monosomy 8 | 10.3 | 5.4 | 18 (2.7%) | 5 (27.8%) | |||
| Trisomy 8 | 35.9 | 7.2 | 24 (3.6%) | 16 (66.7%) | |||
| MYC SV | TTD | 38.3 | 7.3 | 29 (4.4%) | 270 (41.0%) | 22 (75.9%) | 191 (70.7%) |
| Terminal del | 44.1 | 7.5 | 11 (1.7%) | 5 (45.5%) | |||
| Proximal del | 55.9 | 7.8 | 13 (2.0%) | 7 (53.8%) | |||
| Ig translocation | 61.5 | 7.9 | 13 (2.0%) | 9 (69.2%) | |||
| Non-Ig insertion | 68.0 | 8.1 | 65 (9.9%) | 55 (84.6%) | |||
| Non-Ig translocation | 70.8 | 8.1 | 16 (2.4%) | 5 (31.3%) | |||
| Complex del/gain | 75.0 | 8.2 | 30 (4.6%) | 17 (56.7%) | |||
| Ig insertion | 86.0 | 8.4 | 88 (13.4%) | 69 (78.4%) | |||
| Amplification | 144.1 | 9.2 | 5 (0.8%) | 2 (40.0%) | |||
| Total | 33.4 | 7.1 | 658 (100%) | 356 (54.1%) | |||
Note: MYC abnormality subtype, median MYC TPM values, total frequency of the subtype in the CoMMpass cohort (n = 658), and percentage of hyperdiploidy in each MYC subtype. Overall P value (No MYC vs. MYC subtypes) determined by Kruskal–Wallis test. Individual P values determined by a post hoc Dunn test (Supplementary Table S6).
We compared the SVs surrounding only the MYC gene region of the 9 MYC subtypes to other established patterns of SVs recently described in human cancers, including multiple myeloma (Supplementary Table S1; refs. 27, 28, 29). The tandem duplication (TTD) and deletion (terminal and proximal dels) subtypes were described as simple events with a single SV connecting two DNA breaks. Most (n = 26, 89.7%) non-Ig and Ig translocations were categorized as balanced or unbalanced translocation events with no more than 2 chromosome partners. Most (n = 148, 96.7%) non-Ig and Ig insertions were categorized as templated insertions, some being simple and others having evidence of complexity (Supplementary Table S1). The complex del/gains subtype involved non-Ig partners and had the most evidence of a complex rearrangement surrounding the MYC gene. Within this subtype, 14 (46.7%) had ≥10 SVs and these were categorized as chromothripsis similar to the definition used in ref. 29. Of the remaining 16 complex del/gain cases with 3–9 SVs around the MYC region, a single case was classified as having chromoplexy and the remaining 15 cases (50.0%) were termed complex, not otherwise specified. We also identified 4 (4.5%) Ig insertions subtypes as having chromothripsis and a single case as being complex, not otherwise specified (Supplementary Table S1). Three (23.1%) of the Ig translocation subtypes were also described as complex. Amplifications were defined as cases where the estimated copy number of MYC was ≥7N. The 5 MYC amplifications were achieved by either chromothripsis (n = 4) or aneuploidic gain or double minute (n = 1; Supplementary Table S1). Of 270 cases with a MYC SV, the most common SV category was templated insertion (54.8%; Supplementary Table S1). If eliminating simple SVs (TTD and terminal and proximal dels), the frequency of templated insertion was 68.2% of cases, similar to that reported in ref. 29.
Of 365 total MYC SVs identified in 270 cases with a MYC SV, there were 146 unique partner genes (Fig. 1A; Supplementary Table S2; Supplementary Fig. S2A–S2C). Although the most common gene partner involved enhancers of the following genes, IgH at 14q32.33 (13.7%), IgL at 22q11.22 (9.9%), NSMCE2 at 8q24.13 (8.5%), TXNDC5 at 6p24.3 (7.1%), IgK at 2p11.2 (5.5%), FAM46C (TENT5C) at 1p12 (3.0%), and CSMD3 at 8q23.3 (2.2%), recurrent partners were found in association with specific subtypes (Supplementary Table S3). The complex del/gain subtype was enriched for NSMCE2 and CSMD3 partners and the proximal dels involved mostly NSMCE2. Although each Ig insertion case at minimum included IgH, IgL, or IgK, 55.7% of the cases (49/88) had three or more partners (including MYC) in the “templated insertion chain” involving non-Ig genes, the most common being TXNDC5. Of the 49 Ig insertion cases with multiple partners, 8 (16.3%) did not have a direct connection between the Ig and MYC gene regions, but rather were separated by a non-Ig insertion in between (5 IgH, 1 IgK, and 2 IgL). TXNDC5 was the most common MYC partner in the non-Ig insertion subtype. The most common gene partner in non-Ig translocations was FAM46C. Non-Ig insertion subtype was also commonly associated with three or more partners within the “templated insertion chain” (Supplementary Table S3 and S4). No clear MYC partner was identified in TTD and terminal del subtypes. Similar to Mikulasova and colleagues, (12), 57.1% of cases had two chromosome partners, 23.6% had three, 12.1% had four partners, and 7.1% had five or more partners (Supplementary Table S4). In contrast to the high frequency of MYC SVs, only 8 (1.2%) cases had nonsynonymous MYC SNVs with a median MYC expression of 42.9 TPM, with 6 cooccurring in cases that also had a MYC SV (Supplementary Table S5).
Figure 1.

MYC abnormality subtypes, partner genes, and MYC gene expression. A, Bubble plots for MYC SV partners. Gene name (x-axis) is plotted by chromosome (y-axis). No SV involving chromosome Y was detected. SV frequencies are indicated with circle size, and SV group by color (see legend). B, ROC curve for MYC Alteration Classification from MYC expression. ROC curve plotting the false-positive rate versus true-positive rate for the classification of samples for MYC alterations using mRNA expression levels in transcripts per million (TPM). Dotted lines give AUC = 0.5 for an uninformative classifier. C, Boxplot and median MYC transcript levels (log transform of Salmon TPM) of MMRF CoMMpass cases (n = 658), with observed SVs. Significant differences in each subtype were compared with the subtype with no MYC SV (***, P < 0.0001; **, P < 0.001; *, P < 0.05). D, Pie charts showing the distribution of the MYC SV in association with predicted FISH detection.
Type of MYC SV and association with MYC gene expression
We next compared MYC RNA levels in each MYC SV subtype. MYC RNA levels have previously been shown to correlate with the expected MYC gene signature (genes directly targeted by MYC) and a MYC gene signature has also been shown to correlate with MYC protein levels (30, 31). These findings suggest that MYC RNA levels are functionally associated with the predicted effect of MYC transcription. MYC expression correlated with the presence of a MYC SV using ROC curve analysis plotting the false-positive versus true-positive rate for the classification of samples for MYC alterations using MYC TPM data. The AUC value of 0.823 suggests strong correlation of MYC expression with the presence of a MYC SV (Fig. 1B). Significant differences in MYC expression were also observed in some MYC SV subtypes compared with cases without a MYC SV (Fig. 1C; Table 1; Supplementary Table S6). Lowest median MYC expression was observed in cases with a MAX SNV (1.1 TPM) or monosomy 8 (10.3 TPM). The 327 cases with no MYC SV had a median MYC expression of 21.6 TPM. Compared with cases with no MYC SV, expression of MYC was significantly increased in cases with proximal del (55.9 TPM), non-Ig insertion (68.0 TPM), non-Ig translocation (70.8 TPM), complex del/gain (75.0 TPM), Ig insertion (86.0 TPM), and amplification (144.1 TPM) and was significantly reduced in cases with a MAX SNV (Fig. 1C; Table 1; Supplementary Table S6). Significant differences in MYC expression were not identified between cases with no MYC SV and monosomy 8, trisomy 8, TTD, terminal del, and Ig translocations and between many of the individual MYC SV subtypes (Supplementary Table S6).
Type of MYC SV and detection by FISH
Given the heterogeneity of the MYC SVs identified by NGS and our previous observation of reduced detection of MYC SVs by MYC BAP FISH compared with NGS (15), we evaluated which MYC SVs give an abnormal MYC BAP FISH result using our Mayo Clinic cohort with MPseq and FISH data (n = 140). Similar to Abdallah and colleagues (16), 7 cases (5.0%) were abnormal using the MYC BAP FISH probe while 41 cases (29.3%) were abnormal by MPseq. Using MPseq and FISH data described in Smadbeck and colleagues (15), subtypes more likely to be undetected by FISH include proximal deletions and non-Ig and Ig insertions (Supplementary Table S7; Supplementary Fig. S3).
We next analyzed 658 CoMMpass cases with the goal to infer, based on visualization of the genomic architecture from WGS and knowledge of the location of the MYC BAP FISH probes, whether a MYC SV could be detected by FISH. Using this approach, we predicted a MYC SV could be detected by FISH in 82/658 (12.5%) cases, similar to the approximately 13%–15% detected using MYC FISH in previous studies (32, 33). We predicted the MYC BAP FISH probe would miss approximately 69.6% of all MYC SVs and would have reduced sensitivity in detecting TTD, terminal deletions, non-Ig insertions, and Ig insertions (Supplementary Table S8; Fig. 1D). FISH was predicted to detect most amplifications, complex deletion/duplications, most non-Ig and Ig translocations, and some non-Ig and Ig insertions (Supplementary Table S8; Fig. 1D). Of cases with hyperdiploidy, 75.4% had a MYC SV predicted to be missed by FISH. Cases with MYC SVs predicted to be missed by FISH had a slightly lower MYC gene expression (63.7 TPM, range: 5.2–611.5 TPM) compared with cases predicted to be detected by FISH (80.1 TPM, range: 8.9–1,111.8 TPM; P = 0.119; Supplementary Table S8).
MYC gene expression and patient survival
We next evaluated whether elevated MYC gene expression, irrespective of MYC SV status, was associated with differences in patient survival. Boxplot analysis was used to categorize MYC expression in 631 CoMMpass cases with available OS and MYC expression data. Top quartile/high MYC expression was defined as MYC expression ≥75.0 TPM, and bottom quartile/low MYC expression was defined as ≤16.5 TPM. Although mean OS and PFS was shorter in patients with high MYC expression compared with patients with low MYC expression (OS: 4.6 years vs. 5.3 years, P = 0.038; PFS: 2.5 years vs. 3.1 years, P = 0.068; Fig. 2A and B), the findings were only significant for OS. Using MYC expression as a continuous variable, high MYC expression was associated with an increased risk of death on univariate analysis (P = 0.027), but this was not considered significant in a multivariate model including MYC expression and other high-risk abnormalities such as TP53 deletion; 1q gain; high-risk translocations t(4;14), t(14;16) and t(14;20); ISS stage III; and ≥70 years of age (Table 2). Similar observations were observed when analyzing MYC as a categorical variable (high vs. low/medium MYC expression) in the full cohort (Supplementary Table S9). High MYC expression was significantly associated with inferior OS in cases classified as nonhyperdiploid (P = 0.001), but not in cases classified as hyperdiploid (P = 0.843; Supplementary Fig. S4).
Figure 2.
![Figure 2. Impact of MYC gene expression on OS and PFS. A, A comparison of OS (years) in patients with low MYC expression (≤16.5 TPM; blue line), medium MYC expression (16.5–75 TPM; green line), and high MYC expression (≥75.0 TPM; red line) detected by RNAseq from the CoMMpass cohort. OS time (mean) was 5.3 years [95% confidence interval (CI): 5.0–5.7], 4.8 years (95% CI: 4.6–5.1), and 4.6 years (95% CI: 4.2–5.0) in low (n = 158), medium (n = 314), and high (n = 159) MYC expression cohorts, respectively. B, A comparison of PFS (years) in patients with low MYC expression (≤16.5 TPM; blue line), medium MYC expression (16.5–75 TPM; green line), and high MYC expression (≥75.0 TPM; red line) detected by RNAseq from the CoMMpass cohort. PFS time (median) was 3.1 years (95% CI: 2.4–3.9), 2.9 years (95% CI: 2.4–3.3), and 2.5 years (95% CI: 1.7–3.0) in low (n = 158), medium (n = 311), and high (n = 159) MYC expression cohorts, respectively. C, Pie charts showing the distribution of the MYC SV in association with MYC gene expression.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9401535_5430fig2.jpg)
Impact of MYC gene expression on OS and PFS. A, A comparison of OS (years) in patients with low MYC expression (≤16.5 TPM; blue line), medium MYC expression (16.5–75 TPM; green line), and high MYC expression (≥75.0 TPM; red line) detected by RNAseq from the CoMMpass cohort. OS time (mean) was 5.3 years [95% confidence interval (CI): 5.0–5.7], 4.8 years (95% CI: 4.6–5.1), and 4.6 years (95% CI: 4.2–5.0) in low (n = 158), medium (n = 314), and high (n = 159) MYC expression cohorts, respectively. B, A comparison of PFS (years) in patients with low MYC expression (≤16.5 TPM; blue line), medium MYC expression (16.5–75 TPM; green line), and high MYC expression (≥75.0 TPM; red line) detected by RNAseq from the CoMMpass cohort. PFS time (median) was 3.1 years (95% CI: 2.4–3.9), 2.9 years (95% CI: 2.4–3.3), and 2.5 years (95% CI: 1.7–3.0) in low (n = 158), medium (n = 311), and high (n = 159) MYC expression cohorts, respectively. C, Pie charts showing the distribution of the MYC SV in association with MYC gene expression.
Table 2.
The influence of MYC expression, TP53 deletion, 1q gain, high-risk translocations, ISS stage, patient age, and hyperdiploidy on OS and PFS.

Although median age was similar among cases with high MYC expression (64 years) compared with low MYC expression (63 years), cases with high MYC expression had an increased frequency of TP53 deletion (14.5%), 1q gain (48.0%), and ISS stage III (36.0%) compared with cases with low MYC expression (8.0% with TP53 deletion, 24.1% with 1q gain, 25.4% ISS stage III). Low MYC expression was associated with a low frequency of any MYC SV (8.6%), including MYC/IgL SVs (1.1%; Fig. 2C; Supplementary Table S10). Of cases with a MYC SV present in the low MYC expression group, 3.2% were TTD, 2.1% were non-Ig insertion, 0.5% had a complex del/gain, and 1.1% had an Ig insertion. In contrast, 80.2% of cases in the high MYC expression group had evidence of a MYC SV. The most common MYC SV in this group included Ig insertion (33.8%), with 35% of these Ig insertion cases involving IgL. Twenty-six cases (17.2%) with high MYC expression had no MYC abnormality detected (Fig. 2C; Supplementary Table S10).
MYC SVs detected by FISH, but not by NGS, are associated with poor survival outcome
We compared the impact of MYC SVs on patient survival when detected by FISH or NGS. We recently reported that diseased survival was shorter in patients with a MYC SV compared with patients without a MYC SV using the MYC BAP FISH probe (5.3 vs. 8.0 years, P < 0.001; ref. 16). Among patients with hyperdiploidy, OS was decreased in patients with a MYC SV detected by FISH compared with those without a MYC SV (5.7 vs. 8.6 years, P = 0.007; Fig. 3A). Similarly, among patients without hyperdiploidy, OS was decreased in patients with a MYC SV detected by FISH compared with those without a MYC SV (2.8 vs. 6.8 years, P < 0.0001; Fig. 3B). However, among patients with hyperdiploidy, PFS was similar to those with a MYC SV detected by FISH compared with those without a MYC SV (4.4 vs. 4.3 years, P = 0.680; Fig. 3C). In contrast, among patients without hyperdiploidy, PFS was decreased in those with a MYC SV detected by FISH compared with those without a MYC SV (2.6 vs. 3.5 years, P < 0.001; Fig. 3D).
Figure 3.
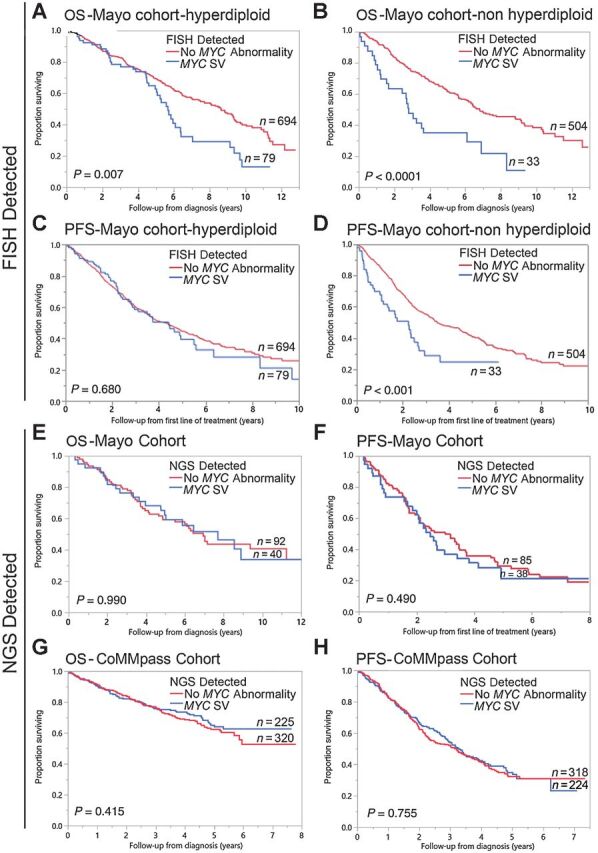
Impact of MYC SV detected by FISH or NGS on OS and PFS. A comparison of OS (years) in patients with a MYC SV detected by FISH (blue curve), and patients without MYC SV (red curve) among patients with hyperdiploidy in A or among patients without hyperdiploidy in B from the Mayo Cohort. OS time (median) in the hyperdiploid group was 5.7 (95% CI: 5.0–6.4) years (n = 79) and 8.6 (95% CI: 7.5–9.3) years (n = 694) in the MYC SV and no MYC SV groups, respectively. OS time (median) in the nonhyperdiploid group was 2.8 (95% CI: 1.5–6.1) years (n = 33) and 6.8 (95% CI: 6.2–8.8) years (n = 504) in the MYC SV and no MYC SV groups, respectively. A comparison of PFS (years) in patients with a MYC SV detected by FISH (blue curve), and patients without MYC SV (red curve) among patients with hyperdiploidy in C or among patients without hyperdiploidy in D from the Mayo Cohort. PFS time (median) in the hyperdiploid group was 4.4 (95% CI: 2.9–5.5) years (n = 79) and 4.3 (95% CI: 3.7–4.9) years (n = 694) in the MYC SV and no MYC SV groups, respectively. PFS time (median) in the nonhyperdiploid group was 2.6 (95% CI: 1.1–2.7) years (n = 33) and 3.5 (95% CI: 3.1–4.5) years (n = 504) in the MYC SV and no MYC SV groups, respectively. E, A comparison of OS (years) in patients with MYC SV detected by MPseq (blue curve) and patients without a MYC SV detected by MPseq (red curve) from the Mayo cohort. OS time (median) was 7.7 (95% CI: 4.8–not reached) years (n = 40) and 6.9 (95% CI: 4.8–11.2) years (n = 92) in the two groups, respectively. F, A comparison of PFS (years) in patients with MYC SV detected by MPseq (blue curve) and patients without MYC SV detected by MPseq (red curve) from the Mayo cohort. PFS time (median) was 2.5 (95% CI: 1.9–3.4) years (n = 38) and 3.0 (95% CI: 2.1–3.5) years (n = 85) in the two groups, respectively. G, A comparison of OS (years) in patients with a MYC SV detected by WGS (blue curve) and patients without a MYC SV detected by WGS (red curve) from the CoMMpass cohort. OS time (mean) was 5.7 (95% CI: 5.3–6.1) years (n = 225) and 5.5 (95% CI: 5.2–5.9) years (n = 320) in the MYC SV and no MYC SV groups, respectively. H, A comparison of PFS (years) in patients with MYC SV detected by WGS (blue curve) and patients without a MYC SV detected by WGS (red curve) from the CoMMpass cohort. PFS time (median) was 3.3 (95% CI: 2.8–3.9) years (n = 224) and 3.1 (95% CI: 2.3–3.7) years (n = 318) in the MYC SV and no MYC SV groups, respectively.
In contrast, there was no significant difference in OS and PFS between patients from the Mayo cohort with a MYC SV detected by MPseq compared with those without a MYC SV (OS: 7.7 vs. 6.9 years, P = 0.990 and PFS: 2.5 and 3.0 years, P = 0.490; Fig. 3E and F). Similarly, there was no significant difference in OS and PFS between patients from the CoMMpass cohort with a MYC SV detected by WGS compared with those without a MYC SV (OS: 5.7 and 5.5 years, P = 0.415; PFS: 3.3 and 3.1 years, P = 0.755; Fig. 3G and H). Although no significant differences in OS and PFS between patients with a MYC SV and those without a MYC SV were observed, a trend toward improved OS and PFS was identified in the hyperdiploid cohort in contrast to the nonhyperdiploid cohort (Supplementary Fig. S5).
Although evaluation of all MYC SVs together resulted in no significant differences in OS and PFS compared with cases with no MYC SVs, the impact of individual MYC SV subtypes on OS and PFS was further evaluated. Among the nine MYC SV subtypes, only non-Ig insertion and Ig insertion subtypes showed significant differences in either OS and/or PFS in univariate analysis (Supplementary Table S11). Patients with non-Ig insertion subtype had improved OS and PFS, while patients with Ig insertion had reduced OS and PFS (Fig. 4; Supplementary Fig. S6). Non-Ig insertion subtype was associated with improved OS [risk ratio (RR) 0.33, P = 0.004] and PFS (RR 0.60, P = 0.018), while Ig insertion subtype was associated with reduced PFS (RR 1.46, P = 0.016) on univariate analysis (Supplementary Tables S12 and S13). Among the Ig insertion subtype, only those involving the IgL gene partner were associated with reduced PFS (RR 2.12, P < 0.001) on univariate analysis (Supplementary Table S14). In a multivariate model including non-Ig insertion and the favorable risk category of hyperdiploidy, non-Ig insertion retained its prognostic value at predicting OS and PFS (OS RR 0.32, P = 0.005; PFS RR 0.60, P = 0.025; Supplementary Table S12). In a multivariate model including either Ig insertion or IgL SVs (all IgL insertions) and other high-risk abnormalities, both Ig insertion and IgL SVs retained their prognostic significance at predicting PFS (Ig insertion, RR 1.40, P = 0.042; IgL, RR 2.29, P < 0.001; Supplementary Tables S13 and S14), with a trend toward reduced OS and PFS in Ig insertion subtypes with increased MYC partners (Supplementary Fig. S7).
Figure 4.

Impact of different MYC SVs detected by NGS on OS and PFS. A, A comparison of OS (years) in patients with no MYC abnormality (no MYC SV, MAX SNV, monosomy 8, and trisomy 8; green line), patients with non-Ig insertion (blue line), patients with IgL rearrangements (all IgL insertions; red line), and all others (orange line) detected by WGS from the CoMMpass cohort. OS time (mean) was 5.5 (95% CI: 5.2–5.9) years (n = 320), 6.9 (95% CI: 6.3–7.4) years (n = 55), 4.3 (95% CI: 3.2–5.3) years (n = 28), and 5.4 (95% CI: 4.9–5.9) years (n = 142) in the no MYC abnormality, non-Ig insertion, IgL, and other group, respectively. B, A comparison of PFS (years) in patients with no MYC abnormality (no MYC SV, MAX SNV, monosomy 8, and trisomy 8; green line), patients with non-Ig insertion (blue line), patients with IgL rearrangements (all Ig insertions; red line), and all others (orange line) detected by WGS from the CoMMpass cohort. PFS time (median) was 3.2 (95% CI: 2.4–3.9) years (n = 318), 4.8 (95% CI: 3.6–6.1) years (n = 55), 1.4 (95% CI: 1.2–1.6) years (n = 28), and 3.1 (95% CI: 2.6–3.5) years (n = 141) in the no MYC abnormality, non-Ig insertion, IgL, and other group, respectively.
Discussion
SVs involving the MYC proto-oncogene are common secondary events in multiple myeloma and are associated with SMM to multiple myeloma disease progression (4–6, 8, 9, 11–14). We observed MYC SVs in 41% of multiple myeloma cases detected by WGS and similar to other studies, WGS identified a greater frequency of MYC SVs compared with FISH (4, 11–13). Among the nine MYC SV subtypes, the non-Ig insertion and Ig insertion subtypes, namely IgL insertions, showed significant differences in outcome in comparison with cases without a MYC SV, similar to other studies (4, 11, 12). The non-Ig insertion subtype was associated with improved outcomes, while the Ig insertion subtype (specifically when partnered with IgL) was associated with poor outcomes. Thus, MYC appears to be associated with poor outcome only when partnered with IgL. The main driver is likely the association with IgL as IgL SVs, even in the absence of partnering with MYC, are associated with poor outcome (11). The poor outcome associated with IgL SVs has been proposed to involve the strong association of the IKZF1 transcription factor to the IgL locus rendering cases with IgL SVs less sensitive to immunomodulatory drug treatment (11). How the non-Ig insertion subtype confers a prognostic benefit independent of hyperdiploidy remains unknown.
Within clinical genomics laboratories, MYC SVs are most often identified using BAP FISH probes spanning the MYC genomic region. FISH detects approximately 13%–15% of MYC SVs in newly diagnosed multiple myeloma (32, 33), compared with approximately 23%–42% detected by WGS (4, 11–13). FISH underestimates the frequency of MYC SVs in multiple myeloma by about 2–3 fold (4, 8, 12, 15), likely due to the heterogeneity of MYC SV breakpoints. This makes the design of FISH probes capable of capturing all MYC SVs a significant challenge. Similar to multiple myeloma, approximately 32% of diffuse large B-cell lymphomas were also discordant between FISH and a hybrid-capture sequencing assay (34). Most of these discordant cases had non-Ig partners with breakpoints outside of the genic cluster (34). Most of the cases with non-Ig SVs represented high-grade B-cell lymphomas, a WHO entity that requires correct MYC classification for accurate diagnosis and patient management. Understanding the limitations of the MYC BAP probe is critical because clinical laboratories use FISH for the evaluation of MYC SVs and evidence of false-negative MYC BAP FISH results have been reported (15, 35–38). We show that FISH has reduced sensitivity toward non-Ig and Ig insertion subtypes because these rearrangements often occur between the 5′ and 3′ MYC FISH probes and do not always result in a separated 5′ and 3′ FISH pattern. Although the MYC FISH probe fails to detect approximately 70% of MYC SVs that are detected by NGS, the MYC probe can still identify MYC SVs in approximately 12% of patients with multiple myeloma. MYC SVs detected by FISH have a higher gene expression and are more likely to be associated with poor outcome suggesting a different pathogenic role compared with other MYC SVs.
Older studies using FISH or target capture–based sequencing found that MYC SVs were associated with inferior survival (7–10). Another study identified MYC SVs in 13% of patients with multiple myeloma with no correlation with other primary abnormalities and no prognostic impact on survival (33). Whether these findings are explained by differences in MYC probe placement or differences in the proportion of hyperdiploidy to nonhyperdiploidy is uncertain. Other studies also found no difference in outcome between cases with or without MYC SVs detected using WGS. Although MYC SVs are more common in hyperdiploidy (4, 7, 9, 11, 12, 14, 26), the influence of MYC SVs on survival in this group has been unclear. While previous studies have shown that FISH-detected MYC SVs are associated with poor outcome only in the hyperdiploid group (7), our findings suggest that FISH-detected MYC SVs and also high MYC expression may be more predictive of poor outcome in the nonhyperdiploid group. It is possible that differences in the frequency and ratio of the favorable non-Ig insertion to the unfavorable Ig insertion subtype between hyperdiploid and nonhyperdiploid groups (Supplementary Fig. S8) may explain some of the differences in outcomes between these cohorts when evaluating MYC SVs.
Previous studies have shown that MYC expression levels correlate with an expected MYC gene signature (30). A MYC signature has also been shown to correlate with MYC protein levels detected by IHC (31), providing support that MYC RNA levels are associated with the predicted effect of MYC transcription. MYC overexpression using IHC has been identified in approximately 40% of patients with multiple myeloma, similar to the frequency of MYC SVs identified in this study, and was associated with reduced survival (39, 40). We also observe that increased MYC expression was independently associated with poorer OS, but primarily in nonhyperdiploid cases. These differences may be explained by the reduced frequency of the non-Ig insertion subtype in the nonhyperdiploid group. We also observed 26 cases (4%) had high MYC expression without evidence of a MYC SV. This finding may be due to epigenetic factors, other SNVs, a MYC SV not detected by NGS, or by increased signaling from a pathway such as NF-κB that promotes MYC expression (41). Elevated BIRC3, NFKB2, and NFKBIA expression was observed in cases with high MYC expression without a MYC SV (data not shown), consistent with a linear relationship between MYC expression and NF-κB index in cases without a MYC SV (4). Although high MYC expression in this subtype with no MYC SV showed a trend toward reduced survival, the sample size was small and results were not considered significant (P = 0.234; data not shown). While expression microarray or RNAseq has been proposed in the evaluation of multiple myeloma (42), this approach has not been uniformly applied into clinical practice and the relative instability of RNA compared with DNA-based genetic tests can be problematic. Evaluation of MYC protein levels by IHC has also not always correlated with the presence of MYC SVs detected by FISH raising the concern that IHC may not be sensitive enough to function as a surrogate for MYC SVs (43), a finding that is not surprising given the challenges of detecting MYC SVs by FISH.
This study is limited by its retrospective nature and by the heterogeneity in treatment regimens in both cohorts. In addition, our sample size for cases with MPseq results was limited; however, the results were consistent with the much larger CoMMpass cohort. Another limitation is the lack of available MYC FISH data and the reliance on inferring the FISH result from our training set that included NGS and FISH data from 70 previous cases from Smadbeck and colleagues (15).
In summary, we show that non-Ig insertion MYC subtypes are associated with improved outcomes and Ig insertion MYC subtypes (specifically those involving IgL) are associated with poor outcomes. Although the MYC BAP FISH approach has the potential to miss nearly 70% of MYC SVs demonstrating that NGS appears to be a more robust technique to characterize a greater fraction of MYC SVs, FISH can identify MYC SVs associated with higher gene expression and poorer outcome. In addition to identifying a greater fraction of MYC SVs, NGS has the potential to identify other clinically significant SVs, CNAs, and SNVs that are currently not routinely evaluated in clinical genomics laboratories. Although NGS is currently associated with increased costs, longer assay turnaround times, greater analysis complexities, and increased data storage requirements compared with FISH (15), NGS approaches should be evaluated as a replacement technique for a more comprehensive evaluation of the tumor clone, in comparison with traditional cytogenetic methodologies such as FISH.
Authors' Disclosures
P.L. Bergsagel reports personal fees from BMS, Janssen, Novartis, Amgen, Pfizer, and Oncopeptides outside the submitted work. L.B. Baughn reports grants from NCI, P50CA186781 during the conduct of the study, as well as grants from NCI, U54 outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Supplemental materials and methods and tables
Supplemental Figure 1 shows diagrams of the predicted genomic architecture of various MYC SVs
Supplemental Figure 2 shows Circos and Bubble plots indicating various MYC partners genes
Supplemental Figure 3 shows schematic diagrams of 5 different MYC SVs and their interphase MYC FISH results
Supplemental Figure 4 shows the influence of high and low MYC gene expression on OS and PFS in either the hyperdiploid or non-hyperdiploid groups of the CoMMpass cohort
Supplemental Figure 5 shows in influence of any MYC SV on OS and PFS in either the hyperdiploid or non-hyperdiploid groups of the CoMMpass cohort
Supplemental Figure 6 shows the influence of various MYC SVs on OS and PFS in either the hyperdiploid or non-hyperdiploid groups of the CoMMpass cohort
Supplemental Figure 7 shows the influence of various MYC SVs on OS and PFS of the CoMMpass cohort
Supplemental Figure 8 shows the distribution of MYC SVs in hyperdiploid and non-hyperdiploid groups
Acknowledgments
This research was supported by the NCI of the NIH (P50CA186781) and from the Marion Schwartz Career Development Award in Multiple Myeloma. The authors thank the MMRF for making the CoMMpass data accessible.
The costs of publication of this article were defrayed, in part, by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
N. Sharma: Data curation, formal analysis, writing–review and editing. J.B. Smadbeck: Data curation, software, formal analysis, writing–review and editing. N. Abdallah: Data curation, software, formal analysis, writing–review and editing. C. Zepeda-Mendoza: Data curation, software, formal analysis, writing–review and editing. M. Binder: Formal analysis. K.E. Pearce: Resources. Y.W. Asmann: Resources. J.F. Peterson: Writing–review and editing. R.P. Ketterling: Writing–review and editing. P.T. Greipp: Writing–review and editing. P.L. Bergsagel: Conceptualization, resources. S.V. Rajkumar: Resources. S.K. Kumar: Resources, writing–review and editing. L.B. Baughn: Conceptualization, data curation, formal analysis, supervision, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol 2020;95:548–67. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 3. Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol 2018;15:409–21. [DOI] [PubMed] [Google Scholar]
- 4. Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020;34:322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A 2000;97:228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiecchio L, Dagrada GP, Protheroe RK, Stockley DM, Smith AG, Orchard KH, et al. Loss of 1p and rearrangement of MYC are associated with progression of smouldering myeloma to myeloma: sequential analysis of a single case. Haematologica 2009;94:1024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weinhold N, Kirn D, Seckinger A, Hielscher T, Granzow M, Bertsch U, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica 2016;101:e116–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, Begum DB, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J 2014;4:e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun 2015;6:6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glitza IC, Lu G, Shah R, Bashir Q, Shah N, Champlin RE, et al. Chromosome 8q24.1/c-MYC abnormality: a marker for high-risk myeloma. Leuk Lymphoma 2015;56:602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun 2019;10:1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mikulasova A, Ashby C, Tytarenko RG, Qu P, Rosenthal A, Dent JA, et al. Microhomology-mediated end joining drives complex rearrangements and overexpression of MYC and PVT1 in multiple myeloma. Haematologica 2020;105:1055–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019;33:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Affer M, Chesi M, Chen WG, Keats JJ, Demchenko YN, Roschke AV, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014;28:1725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smadbeck JB, Peterson JF, Pearce K, Pitel BA, Figueroa AL, Timm M, et al. Mate pair sequencing outperforms fluorescence in situ hybridization in the genomic characterization of multiple myeloma. Blood Cancer J 2019;9:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abdallah N, Baughn LB, Rajkumar SV, Kapoor P, Gertz MA, Dispenzieri A, et al. Implications of MYC rearrangements in newly diagnosed multiple myeloma. Clin Cancer Res 2020;26:6581–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baughn LB, Pearce K, Larson D, Polley MY, Elhaik E, Baird M, et al. Differences in genomic abnormalities among African individuals with monoclonal gammopathies using calculated ancestry. Blood Cancer J 2018;8:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smadbeck JB, Johnson SH, Smoley SA, Gaitatzes A, Drucker TM, Zenka RM, et al. Copy number variant analysis using genome-wide mate-pair sequencing. Genes Chromosomes Cancer 2018;57:459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaitatzes A, Johnson SH, Smadbeck JB, Vasmatzis G. Genome U-Plot: a whole genome visualization. Bioinformatics 2018;34:1629–34. [DOI] [PubMed] [Google Scholar]
- 20. Aypar U, Smoley SA, Pitel BA, Pearce KE, Zenka RM, Vasmatzis G, et al. Mate pair sequencing improves detection of genomic abnormalities in acute myeloid leukemia. Eur J Haematol 2019;102:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heng L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013. Available from: https://arxiv.org/abs/1303.3997. [Google Scholar]
- 22. Karimzadeh M, Ernst C, Kundaje A, Hoffman MM. Umap and bismap: quantifying genome and methylome mappability. Nucleic Acids Res 2018;46:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wickham H. Ggplot2: elegant graphics for data analysis. New York, NY: Springer-Verlag; 2009. p VIII, 213. [Google Scholar]
- 24. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mandrekar JN. Receiver operating characteristic curve in diagnostic test assessment. J Thorac Oncol 2010;5:1315–6. [DOI] [PubMed] [Google Scholar]
- 26. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018;132:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, Tian H, et al. Distinct classes of complex structural variation uncovered across thousands of cancer genome graphs. Cell 2020;183:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rustad EH, Yellapantula VD, Glodzik D, Maclachlan KH, Diamond B, Boyle EM, et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov 2020;1:258–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008;13:167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011;25:1026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001;98:3082–6. [DOI] [PubMed] [Google Scholar]
- 33. Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the intergroupe francophone du myelome. Blood 2007;109:3489–95. [DOI] [PubMed] [Google Scholar]
- 34. Chong LC, Ben-Neriah S, Slack GW, Freeman C, Ennishi D, Mottok A, et al. High-resolution architecture and partner genes of MYC rearrangements in lymphoma with DLBCL morphology. Blood Adv 2018;2:2755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. King RL, McPhail ED, Meyer RG, Vasmatzis G, Pearce K, Smadbeck JB, et al. False-negative rates for MYC fluorescence in situ hybridization probes in B-cell neoplasms. Haematologica 2019;104:e248–e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. May PC, Foot N, Dunn R, Geoghegan H, Neat MJ. Detection of cryptic and variant IGH-MYC rearrangements in high-grade non-Hodgkin's lymphoma by fluorescence in situ hybridization: implications for cytogenetic testing. Cancer Genet Cytogenet 2010;198:71–5. [DOI] [PubMed] [Google Scholar]
- 37. Munoz-Marmol AM, Sanz C, Tapia G, Marginet R, Ariza A, Mate JL. MYC status determination in aggressive B-cell lymphoma: the impact of FISH probe selection. Histopathology 2013;63:418–24. [DOI] [PubMed] [Google Scholar]
- 38. Peterson JF, Pitel BA, Smoley SA, Vasmatzis G, Smadbeck JB, Greipp PT, et al. Elucidating a false-negative MYC break-apart fluorescence in situ hybridization probe study by next-generation sequencing in a patient with high-grade B-cell lymphoma with IGH/MYC and IGH/BCL2 rearrangements. Cold Spring Harb Mol Case Stud 2019;5:a004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szabo AG, Gang AO, Pedersen MO, Poulsen TS, Klausen TW, Norgaard P. Overexpression of c-myc is associated with adverse clinical features and worse overall survival in multiple myeloma. Leuk Lymphoma 2016;57:2526–34. [DOI] [PubMed] [Google Scholar]
- 40. Moller HEH, Preiss BS, Pedersen P, Ostergaard B, Frederiksen M, Abildgaard N, et al. Myc protein overexpression is a feature of progression and adverse prognosis in multiple myeloma. Eur J Haematol 2018 July 12. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 41. Duyao MP, Buckler AJ, Sonenshein GE. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc Natl Acad Sci U S A 1990;87:4727–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Szalat R, Avet-Loiseau H, Munshi NC. Gene expression profiles in myeloma: ready for the real world? Clin Cancer Res 2016;22:5434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nguyen L, Papenhausen P, Shao H. The role of c-MYC in B-cell lymphomas: diagnostic and molecular aspects. Genes 2017;8:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials and methods and tables
Supplemental Figure 1 shows diagrams of the predicted genomic architecture of various MYC SVs
Supplemental Figure 2 shows Circos and Bubble plots indicating various MYC partners genes
Supplemental Figure 3 shows schematic diagrams of 5 different MYC SVs and their interphase MYC FISH results
Supplemental Figure 4 shows the influence of high and low MYC gene expression on OS and PFS in either the hyperdiploid or non-hyperdiploid groups of the CoMMpass cohort
Supplemental Figure 5 shows in influence of any MYC SV on OS and PFS in either the hyperdiploid or non-hyperdiploid groups of the CoMMpass cohort
Supplemental Figure 6 shows the influence of various MYC SVs on OS and PFS in either the hyperdiploid or non-hyperdiploid groups of the CoMMpass cohort
Supplemental Figure 7 shows the influence of various MYC SVs on OS and PFS of the CoMMpass cohort
Supplemental Figure 8 shows the distribution of MYC SVs in hyperdiploid and non-hyperdiploid groups
Data Availability Statement
The MPseq data that support this study have been deposited in the National Center for Biotechnology Information (NCBI)'s Sequence Read Archive with BioProject ID PRJNA739382 and can be accessed at http://www.ncbi.nlm.nih.gov/bioproject/739382. All CoMMpass data used in this study are publicly available [database of Genotypes and Phenotypes (dbGap): phs000748.v1.p1 and EGAS00001001178] and also http://www.ncbi.nlm.nih.gov/bioproject/248538.