

Abstract
Broadly neutralizing antibodies (bNAbs) against HIV-1 develop after prolonged virus and antibody co-evolution. Previous studies showed that sequential immunization with a V3-glycan-patch germline-targeting HIV-1 envelope trimer (Env) followed by variant Envs can reproduce this process in mice carrying V3-glycan bNAb precursor B cells. However, eliciting bNAbs in animals with polyclonal antibody repertoires is more difficult. We used a V3-glycan immunogen multimerized on virus-like particles (VLPs), followed by boosting with increasingly native-like Env-VLPs, to elicit weak broadly neutralizing antibodies in non-human primates (NHPs). Structures of antibody/Env complexes following prime and boost vaccinations demonstrated target epitope recognition with apparent maturation to accommodate glycans. However, we also observed increasing off-target antibodies with boosting. Eight vaccinated NHPs were subsequently challenged with simian-human immunodeficiency virus (SHIV), and seven of eight animals became infected. The single NHP that remained uninfected after viral challenge exhibited one of the lowest neutralization titers against the challenge virus. These results demonstrate that weak heterologous neutralization achieved using sequential immunization is insufficient for protection in this animal model. Thus, improved prime-boost regimens to increase bNAb potency and stimulate other immune protection mechanisms are essential for developing anti-HIV-1 vaccines.
One sentence summary:
Sequential immunization elicits antibodies against the V3-glycan patch of HIV-1 Env in rhesus macaques but also induces off-target antibody responses.
INTRODUCTION
Nearly 40 years after the emergence of the acquired immunodeficiency syndrome (AIDS) pandemic, there is no vaccine or cure. However, pre-clinical models have shown that broadly neutralizing antibodies (bNAbs) against the HIV-1 envelope trimer (Env), the only viral protein on the surface of virions, can prevent HIV-1 infection (1–13), suggesting that a vaccination regimen that elicits bNAbs would be protective. In contrast to most human antibodies, anti-HIV-1 bNAbs are extensively hypermutated (14–17) and develop after prolonged periods of virus and antibody co-evolution in infected humans and non-human primates (NHPs) (18–20). These observations led to the proposal that a vaccine regimen recapitulating this process would require a series of sequential immunogens (21, 22), a hypothesis that was validated in knock-in mice that carried V3-glycan bNAb precursor B cells (23). However, knock-in mice that carry a specific bNAb precursor have a limited B cell antibody repertoire, which is not representative of the complexity of genetically-intact mammalian immune systems.
An obstacle to initiating bNAb responses is that the inferred germline (iGL) precursors of most bNAbs do not bind to HIV-1 envelopes present on circulating HIV-1 strains (24–29). A goal of HIV-1 vaccine development has therefore been to design iGL-targeting immunogens to recruit B cells that express bNAb precursors. Nonetheless, the low frequency of bNAb precursors within a polyclonal repertoire can be circumvented using transgenic iGL knock-in animals.
Previous studies in wild type (wt) animals have shown that initiation of B cell responses depends in part on a specific B cell receptor’s affinity for antigen and on the bNAb precursor frequency (30–34). Thus, protein engineers have focused on producing immunogens that bind with high affinity to specific bNAb precursors. However, optimizing binding to a unique iGL precursor antibody can limit the repertoire of recruited B cells qualitatively and quantitatively. Moreover, it does not take into account observations indicating that when B cells clonally expand within each germinal center, there are multiple founder B cells with a wide range of affinities, and that entry into the germinal center is limited by competition and not absolute affinity (34, 35). To date, no one has consistently elicited heterologously neutralizing serum in wt animals with a polyclonal repertoire, with one exception in which anti-HIV-1 bNAbs were elicited by a fusion peptide-based immunogen in NHPs (36). In that case, however, the elicited antibodies were of limited breadth and potency.
We recently described RC1 and RC1–4fill, low affinity priming Env trimer immunogens engineered to elicit antibody responses that target the V3-glycan patch (37) at the apex of the HIV-1 Env trimer. We showed that immunization of wt animals with RC1–4fill multimerized on virus-like particles (VLPs) elicited antibodies that recognized the V3-glycan patch (38). Here we report the results of boosting experiments in wt animals primed with RC1–4fill-VLPs designed, based in part, on iGL knock-in mouse experiments that succeeded in eliciting bNAbs targeting the V3-glycan patch (23). Although boosting antigens designed to shepherd bNAb development elicited antibody evolution to avoid potential clashes with the gp120 N156 glycan, modified native-like Env gp140 trimer (SOSIP) antigen administration produced off-target responses. Here, we present analyses of post-boost serum and monoclonal antibodies cloned from vaccinated animals, finding both on- and off-target heterologously neutralizing antibodies.
RESULTS
Design of V3-glycan patch immunogens used for sequential immunization
The gp120 V3-glycan patch includes a group of nine high-mannose and complex-type N-glycans surrounding the V3 loop of Env (attached to gp120 residues N133, N137, N156, N295, N301, N332, N339, N385 and N392) (37). Human donor-derived bNAbs targeting this site, including PGT121 (39), 10–1074 (28), and BG18 (40), reach through these glycans using elongated heavy (H) and light (L) chain complementarity-determining region (CDR) loops; in particular, CDRH3 and portions of CDRL1 and CDRL3 contact the highly-conserved Env GDIR motif (G324-D325-I326-R327) in gp120 at the base of V3 (41). 11MUTB includes substitutions in V1 and lacks potential N-linked glycosylation sites (PNGSs) at gp120 positions N133 and N137 (42). We previously described immunogens, RC1 and RC1–4fill, that were derived from a soluble version of the clade A/E BG505 native-like Env trimer (SOSIP.664) (43) and designed to bind germline versions of V3-glycan patch bNAbs. RC1 and RC1–4fill were based on a V3-glycan patch-targeting immunogen 11MUTB (42), which is related to the 10MUT initial immunogen in a series that elicited V3-glycan patch bNAbs in iGL knock-in mice (23).
To construct RC1, we modified 11MUTB to include an additional mutation (N156Q) to remove the N156 glycan, which we reasoned would increase accessibility of V1 residues that interact with V3-glycan patch bNAbs (44, 45) and produce a more electrostatically-neutral Env surface that could facilitate the binding of largely neutral V3-glycan patch iGLs (46). RC1–4fill contains four additional PNGSs at gp120 positions 230, 241, 289 and 344 to cover an immunogenic hole in the BG505 glycan shield (47) with added N-glycans. Prime immunizations with soluble RC1 and RC1–4fill induced V3-glycan specific antibody responses in PGT121/10–1074 iGL knock-in mice and wt mice (38). For other immunizations, both immunogens were multimerized on VLPs derived from the AP205 bacteriophage using the SpyTag–SpyCatcher system (48, 49) to enhance potential avidity effects and limit exposure to off-target epitopes at the Env base (47, 50).
Serum and monoclonal antibodies (mAbs) derived from wt animals immunized with soluble and multimerized RC1-based immunogens were non-neutralizing, as expected after only a prime immunization (38). We therefore designed a boosting regimen for immunized NHPs and rabbits starting with an RC1–4fill-VLP prime (Fig. 1A) to guide neutralizing antibody responses against the V3-glycan patch. For boosting immunizations, purified SOSIP immunogens or “wt” SOSIPs were covalently attached to VLPs (Fig. 1A). To evaluate antibody responses, we constructed HIV-1 pseudoviruses, including strains with RC1 and 11MUTB Envs (table S1), to map targeted epitopes and determine neutralization potencies using an in vitro neutralization assay (51).
Figure 1. NHPs were immunized with a prime and four boosts.
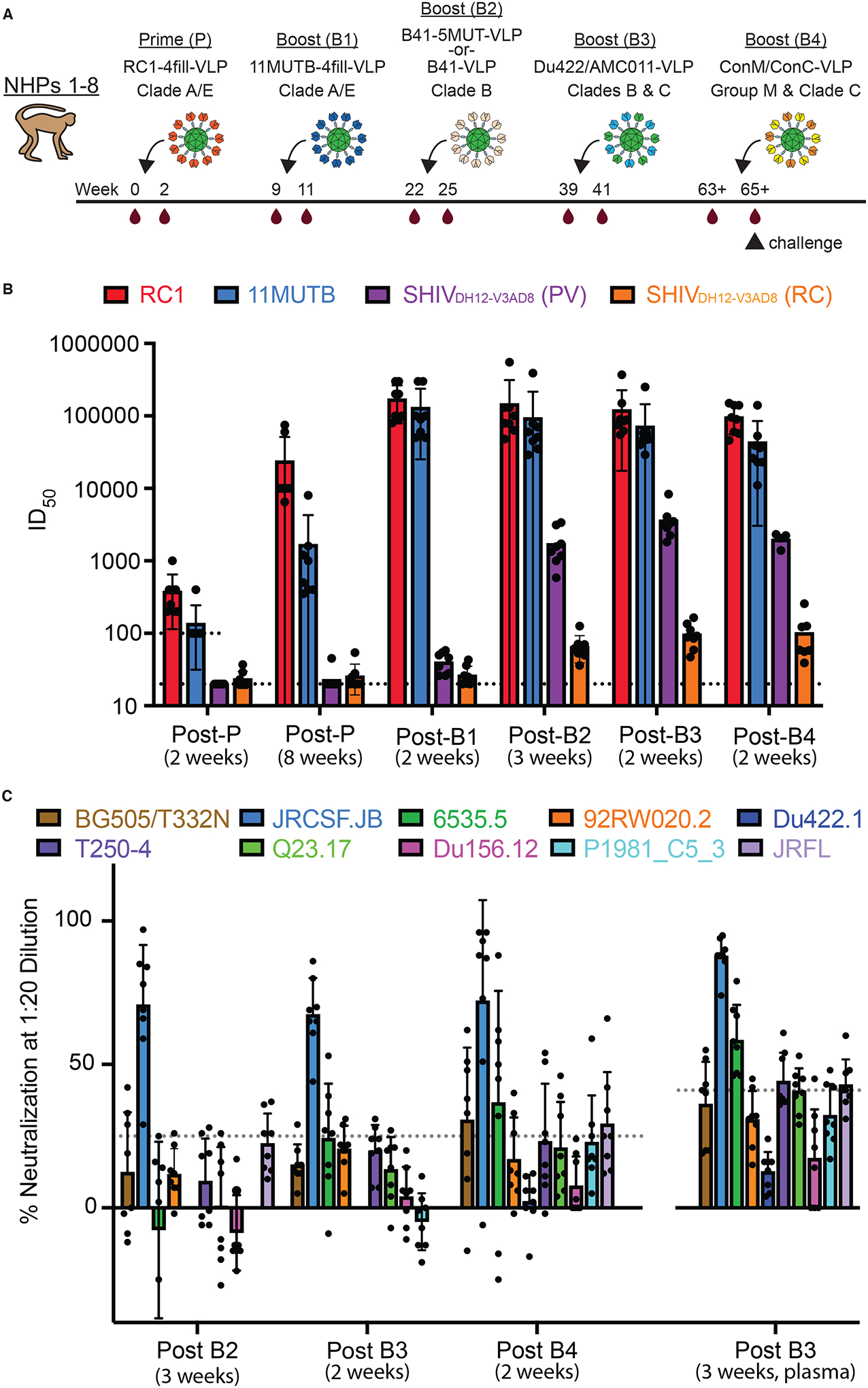
(A) The sequential immunization regimen is shown. NHPs were bled at the indicated times and other times noted in tables S1 and S2. The NHP (NHP 1) used for nsEMPEM (Fig. 2) was immunized with B41–5MUT-VLPs for Boost 2. (B) ID50 values are shown for serum neutralization of viruses at the indicated time points (table S1). Individual dots represent each NHP. The dotted line at 100 indicates the lowest dilution for the 11MUTB and RC1 pseudoviruses. The dotted line at 20 indicates the lowest dilution for SHIVDH12-V3AD8 PV and RC viruses. The arithmetic mean for each entry is indicated by the height of the bar, with the standard deviation shown as a vertical line. PV, pseudovirus; RC, replication competent. (C) Percent neutralization at a 1:20 dilution is shown against heterologous HIV-1 strains at the indicated time points. Individual dots represent each NHP. Samples are serum except for the 3 week post-B3 sample, which was plasma. Dotted lines indicate the highest background neutralization value for the murine leukemia virus (MuLV) control for serum samples (25%; all samples except for post B3, 3 weeks) and plasma samples (41%; post B3, 3 weeks) (table S2). The arithmetic mean for each entry is indicated by the height of the bar, with the standard deviation shown as a vertical line.
Antibody-RC1 structures solved using Fabs from mAbs isolated after the prime suggested potential clashes with the gp120 N156 glycan when present on an HIV Env (38). Thus 11MUTB, which includes the N156 glycan, was a logical choice for the first boosting immunogen. We attached a SpyTagged version of 11MUTB (42) with 4-fill mutations (38) to VLPs to create 11MUTB-4fill-VLPs, which were used for Boost 1 (B1) (Fig. 1A). The clade A/E BG505-based 5MUT immunogen (42) was the third in a series of antigens that elicited bNAbs in immunized knock-in mice (23). For Boost 2 (B2), we used this immunogen, but attempted to avoid BG505 strain-specific responses by engineering the clade B B41 SOSIP trimer (52) to include the 5MUT mutations (42) and an introduced PNGS at gp120 position 289. SpyTagged versions of B41–5MUT and wt B41 were attached to VLPs to create B41–5MUT-VLPs and B41wt-VLPs, respectively, which were used for Boost 2 in two cohorts of four NHPs each (table S1, table S2). For Boost 3 (B3), we diverged from the knock-in mouse experimental protocol to try to increase breadth and further avoid strain-specific responses by making mosaic VLPs to which we coupled two different wt SOSIPs from clade B (AMC011) (53) and clade C (Du422) (54) (AMC011/Du422-VLPs) (Fig. 1A). These Envs were chosen because their native glycan shields cover immunodominant regions of HIV-1 Env, and the corresponding viruses are highly susceptible to neutralization by V3-glycan patch-specific bNAbs (28, 53). An additional shielding glycan was added to AMC011 by introducing a PNGS at gp120 position 230. For Boost 4 (B4), we attempted to further diversify the V3-glycan patch epitope presented on Env trimers, seeking to avoid strain-specific epitopes by constructing a mosaic VLP that was conjugated with consensus group M and consensus clade C SOSIPs (ConM/ConC-VLPs) (55, 56) (Fig. 1A).
Serum samples from prime-boosted wt animals targeted the V3-glycan patch and showed weak, heterologous neutralizing activity
To map epitope(s) recognized by serum antibodies elicited in NHPs, we used an automated competition ELISA in which a randomly biotinylated RC1 trimer was immobilized on an ELISA plate, incubated with a Fab from a potential competitor mAb of known epitope on Env trimer, and probed with serum containing a polyclonal mixture of antibodies (fig. S1A). Controls involved using monoclonal IgG antibodies of known epitopes instead of NHP serum. The following competitor Fabs or control IgG antibodies were used: 10–1074 and PGT128, representative V3-glycan patch bNAbs (28, 39); PGT145 and PG16, V1V2 bNAb (57, 58); 3BNC117 and IOMA, CD4bs bNAbs (26, 45); SF12, a bNAb against the glycan-dominated silent face (59); VRC34, a gp41 fusion peptide-directed bNAb (60), 8ANC195, a bNAb against the gp120-g41 interface (61); 3BC315, a bNAb that binds between adjacent gp41 protomers and neutralizes by promoting Env trimer decay (62); and 10A and 12N, non-neutralizing antibodies that bind to distracting epitopes on BG505 Env: 10A binds to the glycan hole surrounding gp120 residue 241, and 12N binds to the base of trimer base (47).
Results from the competition ELISAs were fairly uniform across the samples from NHPs 1 to 8, showing competition with the V3-glycan patch bNAbs 10–1074 and PGT128 after the prime (fig. S1A). The amount of competition with the V3-glycan patch bNAbs diminished with boosting, especially for the PGT128 competition (fig. S1A). Although these results indicate targeting of the desired epitope, we did not observe 100% competition with 10–1074/PGT128 for any of the samples, consistent with elicitation of antibodies that recognize off-target Env epitopes. Off-target responses increased with boosting as evidenced by increased serum responses against RC1 and proportionally decreased V3-glycan patch binding (fig. S1B).
We also evaluated post-prime and post-boost serum samples by neutralization assays against RC1, 11MUTB, SHIVDH12-V3AD8, and SHIVAD8EO pseudoviruses (PV) (51) and against a replication-competent (RC) SHIVDH12-V3AD8 constructed as described (63) (Fig. 1B, table S1). The RC1 and 11MUTB pseudoviruses represent autologous strains for our immunogens (RC1 for prime and boost immunizations; 11MUTB for boost immunizations). The heterologous Tier 2 SHIVDH12-V3AD8 and SHIVAD8EO viruses were chosen to assess responses against potential viral strains that could be used in later challenge experiments, with SHIVAD8EO representing a highly pathogenic strain (64) and SHIVDH12-V3AD8 being a SHIVAD8EO derivative with less pathogenicity in NHPs (63).
Two weeks after the prime, serum samples from NHPs 1 to 8 exhibited autologous neutralization against RC1 ranging from 50% inhibitory doses (ID50s) of 200 to 1,000, and undetectable neutralization of 11MUTB, SHIVDH12-V3AD8, and SHIVAD8EO except for neutralization of 11MUTB in NHP 4 serum (ID50 = 400) (table S1). By 8 weeks post-prime, autologous neutralization of RC1 had risen to ID50s of 6,500 to 75,000, and responses to 11MUTB ranged from ID50s of 350 to 8000, demonstrating neutralization of a pseudovirus containing the N156 glycan. Concurrently, we observed low titers of heterologous neutralization of SHIVDH12-V3AD8 pseudovirus or replication-competent viruses (ID50s <60) in three of the primed NHPs (table S1).
Three weeks after the first boost with 11MUTB-4fill-VLPs, autologous neutralization of RC1 rose again to ID50s of 80,000 to 300,000 and 11MUTB titers also rose to ID50s of 50,000 to 300,000 (Fig. 1B, table S1). At this time point, titers against SHIVDH12-V3AD8 pseudovirus and replication-competent viruses were barely detectable in all 8 NHPs. By 3 weeks post-boost 2 with B41 wt-VLPs or B41–5MUT-VLPs, neutralization potencies against RC1 and 11MUTB remained high, but titers against the heterologous SHIVDH12-V3AD8 pseudovirus rose to values ranging from 584 to 3378, with detectable neutralization against the replication-competent SHIVDH12-V3AD8 in all 8 NHPs (ID50s from 36 to 92). The mean titers after Boost 2 against the SHIVDH12-V3AD8 pseudovirus were significantly higher in the four NHPs that received the B41–5MUT-VLP boost (NHPs 1,2,5,6) compared with the four NHPs that received the B41wt-VLP boost (NHPs 3,4,7,8) (mean ID50 = 2445 ± 809 versus 1027 ± 283; p = 0.046). We conclude that that the immunization series starting with RC1–4fill-VLPs, followed by 11MUTB-4fill-VLPs and B41–5MUT-VLPs, elicited antibodies that neutralized autologous strains and also exhibited weak activity against a heterologous SHIV pseudovirus.
Two weeks after boost 3, we observed relatively steady titers against RC1 and 11MUTB. Importantly, serum neutralization titers against pseudotyped SHIVDH12-V3AD8 rose to values above 2000 in 7 of 8 animals (mean ID50 = 3671 ± 2004) with an ID50 greater than 8000 observed in NHP 4 (table S1). Similarly, the titers against replication-competent SHIVDH12-V3AD8 rose in most of the NHPs and ranged from 47 to 164, with NHP 4 again exhibiting the highest titer. Samples collected two weeks after boost 4 showed relatively steady neutralization titers in most NHPs against RC1, 11MUTB, and the SHIVDH12-V3AD8 pseudovirus. Titers against replication-competent SHIVDH12-V3AD8 rose or fell in NHPs 1 to 8, with a range from 39 to 256. Not surprisingly, no serum neutralization activity was ever detected against SHIVAD8EO, as this virus is less sensitive than SHIVDH12-V3AD8 to PGT121, a V3-targeting bNAb (63). Overall, the trends across NHPs 1 to 8 (Fig. 1B) suggested that (i) neutralization titers against autologous RC1 and 11MUTB pseudotype viruses increased after the prime and first boost, remaining relatively constant after the remaining boosts with heterologous immunogens, (ii) neutralization titers against the heterologous SHIVDH12-V3AD8 pseudovirus remained at baseline until after Boost 1, rose after Boost 2 and Boost 3, but then remained relatively constant, and (iii) neutralization titers against the replication-competent SHIVDH12-V3AD8 virus were lower than titers against the homologous pseudotype viruses (65, 66).
We next evaluated serum neutralization in immunized NHPs against a panel of heterologous Tier 1 and Tier 2 pseudotyped viruses (Fig. 1C, table S2). Neutralization against these viruses was very weak or undetectable after the prime and Boost 1. Sequential boosting with B41wt-VLPs or B41–5MUT-VLPs elicited weakly neutralizing activity against the heterologous HIV-1 strain JRCSF.JB (Boost 2; table S2). The heterologous neutralizing responses were maintained, or in some cases improved, by the boost of Du422/AMC011-VLPs (Boost 3), but were reduced after the ConC/ConM-VLP boost (Boost 4) (Fig. 1C, table S2). Serum neutralization assays for rabbits immunized with the same prime-boost regimen as the NHPs (fig. S2A) showed qualitatively similar results. No neutralization against heterologous viruses was observed after the prime or Boost 1, weak neutralization against JRCSF.JB was observed after Boost 2 and Boost 3, and no neutralization was observed after the ConC/ConM-VLP boost (fig. S2B).
Polyclonal negative-stain EM demonstrated antibody targeting of the V3-glycan patch
We also used negative stain polyclonal electron microscopy (nsEMPEM) (50, 67) to map the epitopes of elicited antibodies on HIV-1 Envs. In this method, polyclonal Fabs that bind to an antigen are purified from unbound Fabs by size-exclusion chromatography (SEC), imaged by EM, and antibody epitopes are identified in 2D class averages or 3D classes. A caveat of nsEMPEM is that some antibody epitopes in a polyclonal mixture are not visualized in 2D or 3D classes. For example, nsEMPEM mapping of epitopes in plasmas from coronavirus disease 2019 (COVID-19) convalescent donors revealed less epitope diversity than apparent from single B cell cloning of antibody genes or competition experiments (68). We therefore refer to the nsEMPEM-visualized epitopes as “predominant” or “primary” epitopes and acknowledge that other epitopes may exist.
After demonstrating that RC1–4fill-VLP priming elicited similar RC1 and 11MUTB pseudotype virus neutralizing responses in rabbits and NHPs (fig. S2C), we did initial nsEMPEM studies to evaluate polyclonal responses in a rabbit for which we had serum samples in sufficient quantities for Fab isolations after the same prime/boosting regimen used in NHPs (Fig. 1A; fig. S2A; table S3). 3D reconstructions of RC1 bound to polyclonal Fabs derived from rabbit serum after the prime and Boosts 1–3 showed a primary epitope in the vicinity of the V3-glycan patch of the RC1 trimer after a priming immunization (Fig. 2A), demonstrating targeting of the desired Env epitope. In addition, Fabs that target the base of the trimer were identified in all 2D class averages, with their prevalence gradually increasing after each boost (Fig. 2A, fig. S3). Trimer base-binding Fabs also appeared in concurrence with the observation of dissociated Env trimers (fig. S3, fig. S4A), consistent with observations by others (69). In contrast to previous EMPEM analyses of serum from BG505-immunized animals (50, 67), we did not observe Fabs binding to the region of the residue 241 “glycan hole” in serum Fab complexes with RC1 Env trimer (Fig. 2A and B; fig. S3). This indicates that the 4-fill mutations designed to block access to this site in BG505-derived immunogens (38) prevented detectable elicitation of these types of non-neutralizing antibodies.
Figure 2. nsEMPEM shows predominant V3 targeting after the prime and initial boosts.
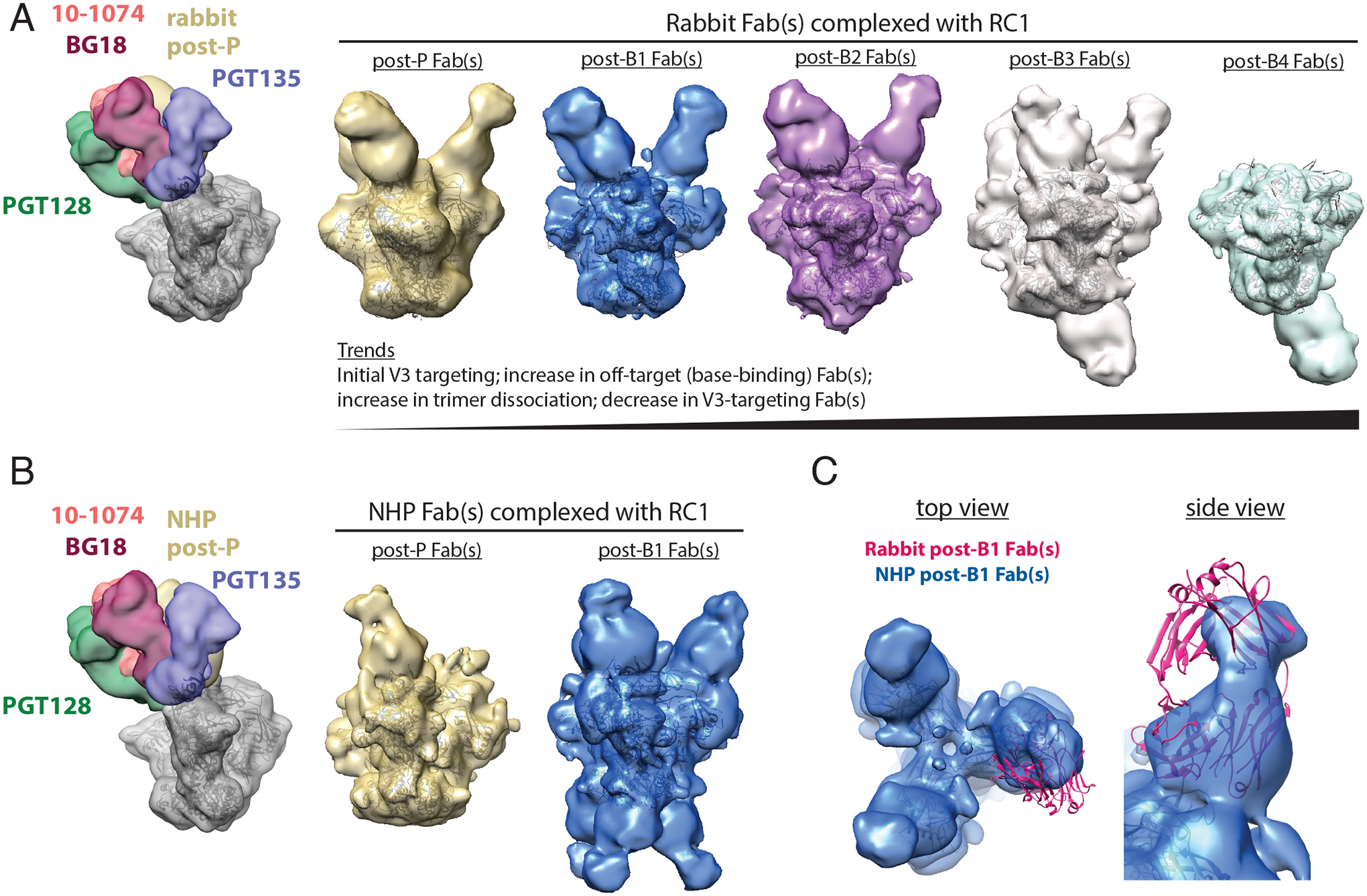
(A) Left: A low-pass filtered structure of representative HIV-1 SOSIP Env trimer is shown complexed with V3-glycan patch bNAb Fabs (PDB codes 5T3Z, 6CH8, 5C7K, 4JM2; colors as indicated) and post-P rabbit Fabs (gold) bound to one Env protomer. Polyclonal Fabs were isolated from Rabbit 2249 (fig. S2B), which received B41–5MUT-VLPs as Boost 2. Right 5 panels: 3D reconstructions of Fabs from antibodies isolated from a rabbit 2 weeks after P, B1, B2, B3, and B4 in complex with RC1 are shown. The predominant states for each time point are shown. See also fig. S3 and S5 for 2D classes, including bottom-binding Fabs after P, B1, and B2 and Fabs bound to dissociated protomers. Coordinates of RC1 (PDB 6ORN) were independently docked into EM density maps. (B) Left: A low-pass filtered structure of representative HIV-1 SOSIP Env trimer is shown complexed with V3-glycan patch bNAb Fabs (colors as indicated) and post-P NHP Fabs (gold) bound to one protomer. Polyclonal Fabs were isolated from NHP 1, which received B41–5MUT-VLPs as Boost 2 (Fig. 1A). Right: 3D reconstructions of Fabs from antibodies isolated from an immunized NHP 2 weeks after P and B1 are shown. See also fig. S4 for 2D classes showing bottom-binding Fabs. (C) Comparison of Fabs binding poses for rabbit and NHP nsEMPEM reconstructions are shown. Rabbit post-B1 Fabs are shown as a pink cartoon of PDB 5UD9 that was docked into the nsEMPEM 3D reconstruction; NHP post-B1 Fabs indicated by the density from the 3D reconstruction.
We next employed nsEMPEM to evaluate serum from NHP 1 (immunized using the regimen that included B41–5MUT-VLPs as Boost 2) after the prime and the first boost, time points for which we had sufficient NHP serum for Fab isolation. After the prime, we observed V3-targeting Fabs, with few to no detectable base-binding Fabs (Fig. 2B; fig. S4B). After Boost 1 with 11MUTB-4fill, we observed an increase in Fabs that targeted both the V3 region and the trimer base (Fig. 2B; fig. S4B). As also observed in the rabbit serum nsEMPEM results, we did not find detectable anti-glycan hole antibodies in 3D reconstructions (Fig. 2B) or 2D class averages of serum Fabs bound to RC1 (fig. S4B).
It is noteworthy that the nsEMPEM results for the NHP serum Fabs, as well as rabbit serum Fabs, showed a primary epitope in the V3 region (Fig. 2A to C), demonstrating successful focusing of the anti-Env antibody response to the desired epitope in two different species of immunized wt animals. Comparison of the binding poses of the V3-directed rabbit and NHP Fabs showed similar V3 targeting, but different orientations of the Fab VH-VL domains (Fig. 2C). In addition, the trimer base-binding Fabs observed in serum from NHP 1 exhibited different characteristics than those boosted in the rabbit: the NHP base-binding Fabs adopted a distinct binding orientation compared to the rabbit Fabs and the predominant class appeared to bind three per trimer, versus one per trimer for the rabbit Fabs (Fig. 2A and B).
The binding pose adopted by the rabbit Fabs after the prime was distinct from the binding poses of V3-glycan patch bNAb Fabs and from the poses of mAbs cloned after the RC1–4fill-VLP prime in wt mice (Ab275mur) and NHPs (Ab874NHP and Ab897NHP) (38) (Fig. 3A and B). However, a new cryo-electron microscopy (cryo-EM) structure (fig. S5, fig. S6; table S4) of an RC1 complex with the prime-elicited mAb Ab1170NHP (38) showed that the Ab1170NHP Fab matched the rabbit nsEMPEM Fabs binding pose more closely (Fig. 3B), providing a higher-resolution structure resembling the Fabs revealed by nsEMPEM. In addition, although a distinct pose, our previous structure of RC1 complexed with Ab275mur also showed a Fab binding pose more similar to the rabbit Fabs (Fig. 3B).
Figure 3. Elicited antibodies adopt binding poses similar to V3 bNAbs.
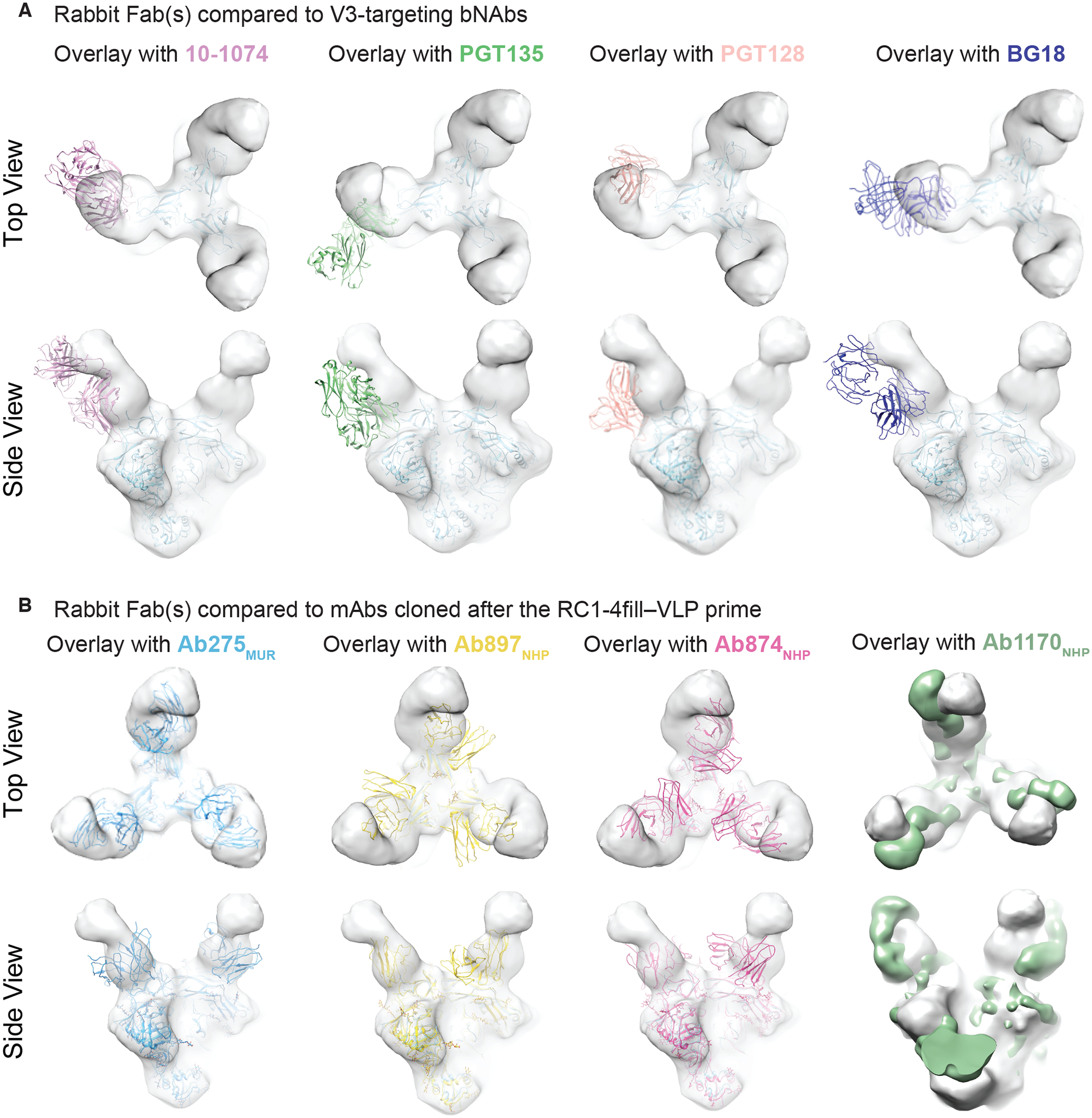
(A and B) EM density maps for 3D reconstruction of RC1 bound to rabbit Fabs elicited after the RC1–4fill-VLP prime are overlaid with coordinates for the indicated bNAbs (PDB codes 5T3Z, 6CH8, 5C7K, 4JM2) aligned on Env trimer (A) or overlaid with coordinates for mAbs elicited from a primed wt mouse (Ab275mur; PDB 6ORQ), a boosted wt mouse (Ab283mur), or a primed NHP (Ab874NHP; PDB 6ORO, and Ab1170NHP; this study, shown as an EM density map) (B).
We extended our previous analysis of the Ab275mur-RC1 cryo-EM structure (PDB 6ORQ) to include a comparison with a new structure of RC1 complexed with Ab283mur (Fig. 4A; fig. S5, table S4). Ab283mur was isolated after an RC1 prime followed by sequential boosting in a wt mouse. After aligning on the RC1 trimer coordinates of both structures, we observed a shift in the mAb pose in a direction that would better accommodate the gp120 N156 glycan, which was present on the boosting immunogen, but not the priming immunogen (Fig. 4B).
Figure 4. Cryo-EM and nsEMPEM Fab-Env structures show apparent accommodation of gp120 N156 glycan after boosting.
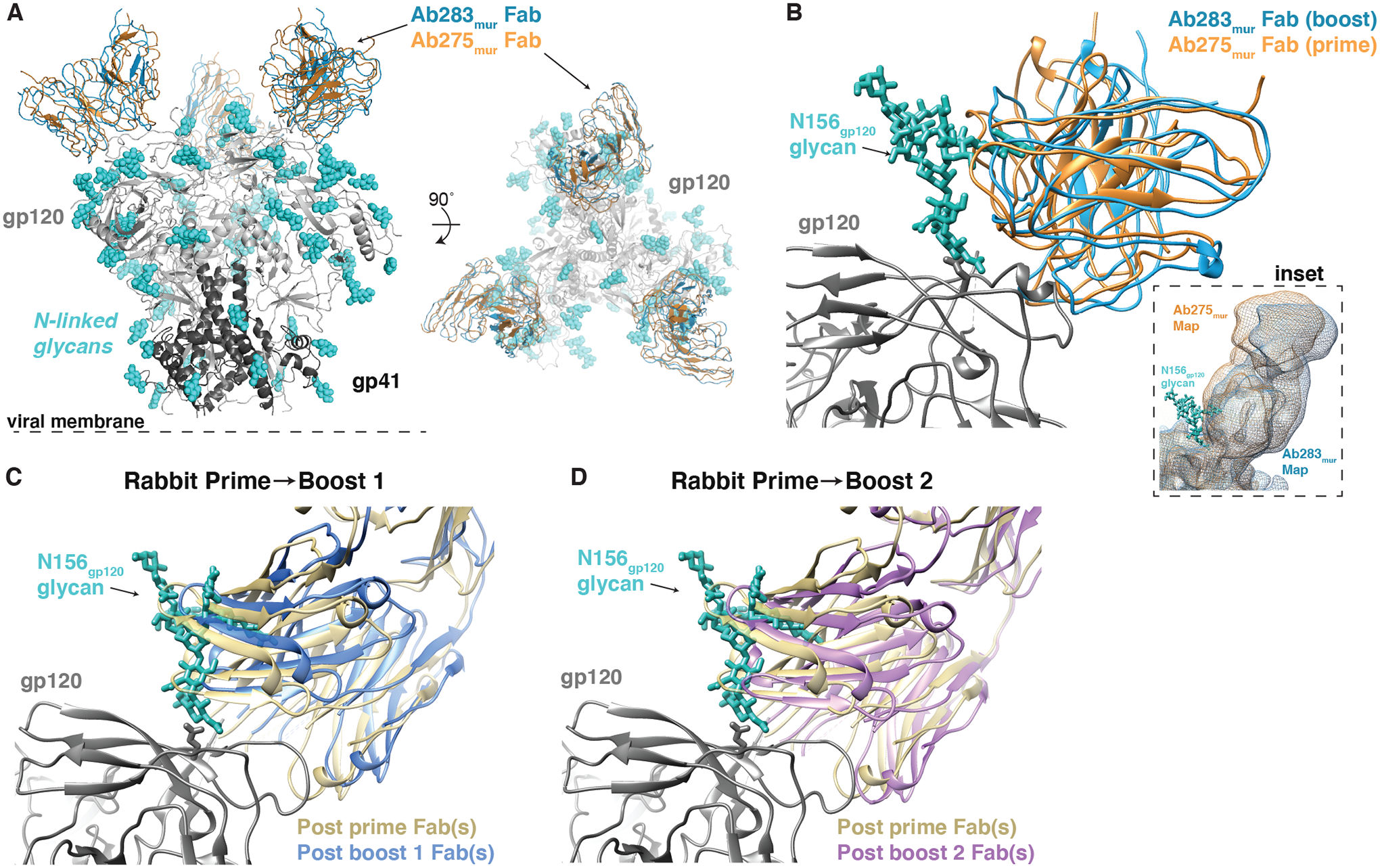
(A) A comparison of Ab275mur-RC1 (PDB 6ORQ) and Ab283mur-RC1 structures is shown. (B) A comparison of binding poses of two mouse monoclonal antibodies, Ab275mur and Ab283mur, isolated from wt mice after a prime only (Ab275mur) or prime plus sequential boost (Ab283mur) is shown. After aligning the Ab283mur-RC1 structure reported here and the Ab275mur-RC1 structure (PDB 6ORQ) on the RC1 coordinates, the pose of Ab283mur was shifted relative to that of Ab275mur in a direction that might better accommodate the gp120 N156 glycan (blue sticks from PDB 5T3X), which was present on boosting immunogens but not the priming immunogen. Inset: Overlay of low-pass filtered single-particle cryo-EM maps of the Fab-RC1 structures. (C and D) Coordinates docked into Fabs-Env nsEMPEM reconstructions (Fig. 2A) (C3 symmetry constraints applied) were aligned on the RC1 trimer and displayed as a close-up view of the antibody interaction site with the VH-VL domains of the post-P and post-B1 Fabs (C) or the post-P and post-B2 Fabs (D). The post-B1 Fabs and post-B2 Fabs appear to shift in a direction that might better accommodate the gp120 N156 glycan (blue sticks from PDB 5T3X).
To address whether the V3-targeting Fabs revealed by nsEMPEM after the prime and first two boosts in rabbits also underwent a shift to accommodate the gp120 N156 glycan, we examined the orientations of the Fabs after aligning on RC1 coordinates fit to the trimer densities. Indeed, Fab coordinates fit to the densities after Boosts 1 and 2 (11MUTB-4fill-VLPs and B41–5MUT-VLPs, respectively) indicated a slightly different binding pose relative to the V3-glycan patch compared with the predominant Fabs after the prime, with the Fabs after boosting being translated in a direction that might more easily accommodate the gp120 N156 glycan (Fig. 4C and D).
We conclude from these structural studies that, as designed, the RC1–4fill-VLP immunogen elicited V3-targeting antibodies. In addition, the initial boosting immunogens elicited antibodies that can better accommodate the gp120 N156 glycan, a potential roadblock in our vaccination regimen. However, the results also demonstrated that the immunization regimen requires further optimization to reduce the elicitation of off-target antibodies that increased upon boosting.
EM of RC1–4fill-VLPs revealed incomplete conjugation and free Env trimers
The observation of increased antibodies against the Env trimer base (Fig. 2A and B; fig. S3, S5B) after boosting suggested accessibility of the SOSIP base to antibodies. To investigate the mechanism by which antibodies could gain access to the Env base in a covalently-coupled VLP in which the trimer base should be inaccessible, we examined SOSIP-coupled VLPs by negative stain EM (nsEM). These imaging studies revealed two relevant findings. First, SpyTagged RC1, but not SpyTagged RC1–4fill, conjugated efficiently to VLPs, as revealed by densities corresponding to closely-packed trimers on RC1-VLPs (fig. S7A, second and third images from left) compared with the relatively sparse trimer densities for RC1–4fill-VLPs (fig. S7A, left). Notably, the epitope for the anti-trimer base antibody 12N (47) was not accessible on densely-packed RC1-VLPs (fig. S7B). As assessed biochemically, with the exception of RC1–4fill, all SpyTagged SOSIP Env trimers, including 11MUTB-4fill, conjugated efficiently to VLPs (fig. S7C). Thus the RC1–4fill immunogen trimer exhibited unique undesirable properties compared to the other immunogen-VLPs. Second, although the SEC protocol efficiently separated free SOSIP trimers from VLP-coupled SOSIP trimers when evaluated at relatively small scales (fig. S7D), the same protocol did not completely isolate free SOSIP trimers when scaled up to make the quantities of SOSIP-VLPs required for NHP and rabbit immunizations (fig. S7E, black trace). Thus the RC1–4fill-VLPs used for the primes in NHPs and rabbits (38) and the SOSIP-VLPs used for Boosts 1 to 3 (table S1, table S2) included free Env trimers in addition to VLP-conjugated trimers. We worked out an alternative SEC protocol for separating SOSIP-VLPs from free trimers in time for Boost 4 (fig. S7E, red trace). These characterizations revealed two potential reasons to explain the presence of the off-target, anti-trimer base antibodies: (i) sparse conjugation of the priming immunogen RC1–4fill, and (ii) the presence of unconjugated Env trimers in the prime and first three boosts.
Single B cell cloning revealed heterologous weakly neutralizing antibodies with epitopes that competed with V3-glycan patch bNAbs and other bNAbs
Env-specific B cells were isolated using a multi bait strategy including Avitagged-biotinylated RC1-glycanKO (38) or an irrelevant, non-HIV-1-based, biotinylated bait and two or three Avitagged-biotinylated SOSIP Env-based baits: RC1, BG505 and B41 (20, 70) (Fig. 5). We screened about 30 IgG monoclonal antibodies (mAb) produced from these sequences against a panel of seven Tier 1 and Tier 2 HIV-1 pseudoviruses (screening panel, Fig. 5A), finding nine that neutralized four or more strains (Fig. 5A; table S4). These were further evaluated against 12 strains from a global HIV-1 reference panel (71) (12-strain panel; Fig. 5A). Although most of the 50% inhibitory concentrations (IC50 values) indicated extremely low potencies, the nine mAbs showed neutralization breadth across this 19-virus panel. In particular, selected mAbs neutralized the heterologous SHIVDH12-V3AD8 pseudovirus with IC50s of <0.020 μg/mL (Ab1456, Ab1457, Ab1415, and Ab1461); these mAbs also exhibited IC50s of ≤1.1 μg/mL against replication-competent SHIVDH12-V3AD8 (Fig. 5A).
Figure 5. mAbs isolated from prime-boosted NHPs show weak, but heterologous, neutralization and target epitopes that compete with V3-glycan patch bNAbs.
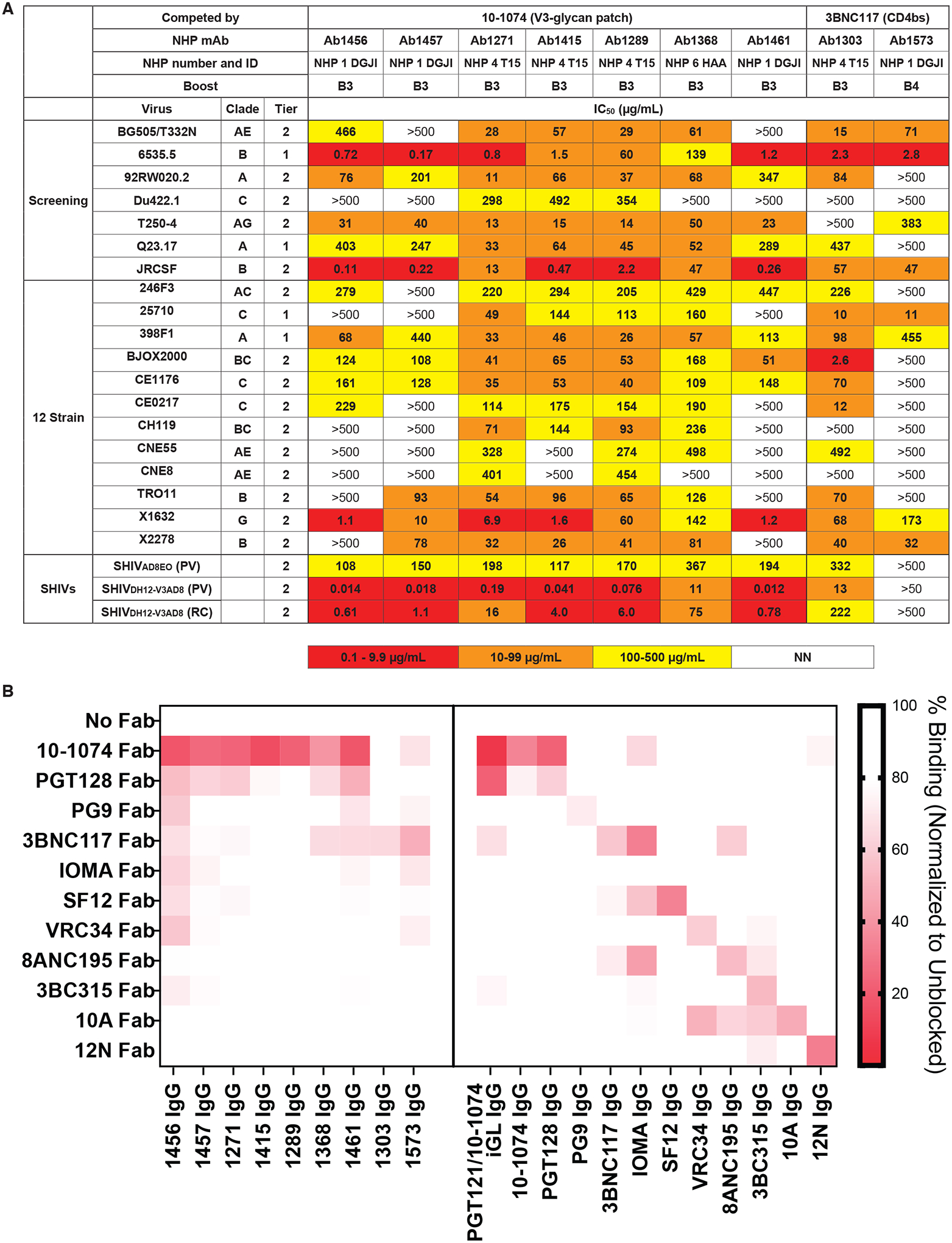
(A) IC50 values against the indicated pseudoviruses are shown for 9 selected mAbs cloned from immunized NHPs for neutralization. NN, non-neutralizing. (B) The heat map shows results of competition ELISAs to map epitopes of mAbs derived from immunized NHPs. Results represent one experiment of two independent replicates. Randomly biotinylated RC1 was bound to a Streptavidin ELISA plate. Each well was then incubated with a potential competitor Fab for 2 hours and then an IgG was added. Bound IgG was detected using an anti-Fc antiserum. The percent binding observed after addition of a competitor Fab was normalized to the binding in the absence of a competitor Fab. NHP IgG results are shown on the left; control bNAb or non-neutralizing IgG antibody (10A and 12N) results are shown on the right.
To map epitope(s) recognized by the NHP mAbs, we adapted the competition ELISA for mAb epitope mapping. For these experiments, we incubated immobilized RC1 with a Fab from a potential competitor mAb and then probed with an NHP IgG or control IgG (Fig. 5B). We used the same competitor Fabs and control IgG antibodies as in the serum competition assay with the addition of PGT121/10–1074 iGL, the inferred germline sequence for the PGT121 and 10–1074 V3-glycan patch bNAbs (28), and the substitution of PG9 (58) as the representative V1V2 bNAb.
Results from the competition ELISA (Fig. 5B) mapped seven mAbs (Ab1456, Ab1457, Ab1271, Ab1415, Ab1289, Ab1368, and Ab1461) as targeting epitopes on Env that were blocked by the V3-glycan patch bNAbs 10–1074 and PGT128 (28, 39) and two mAbs (Ab1303 and Ab1573) as targeting a CD4bs epitope blocked by 3BNC117 (26) (Fig. 5B). Verification of the competition ELISA methodology was provided experiments using control bNAbs targeting known HIV-1 Env epitopes (Fig. 5B). Interesting, although its epitope was not intentionally targeted in our prime-boost protocol, the CD4bs Ab1303 exhibited neutralization activity against almost every strain it was tested against (Fig. 5A), demonstrating that our prime-boost regimen elicited neutralizing antibodies against more than one epitope on HIV-1 Env.
SHIV infection was observed after challenge in 7 of 8 of immunized NHPs
Using a “high dose” challenge regimen (Fig. 6A), four immunized macaques (NHPs 1 to 4) were challenged a single time intrarectally (IR) with 1000 Median Tissue Culture Infectious Dose (TCID50) of SHIVDH12-V3AD8 and 3 of the 4 became infected (Fig. 6B), including NHP 4 (T15), which had generated the highest anti-virus neutralization titers (1:9953) after the 4th boost in the pseudotyped virus assay (table S1). By contrast, NHP 1 (DGJI), which did not become infected after the challenge, showed the lowest SHIVDH12-V3AD8 pseudovirus neutralization titers (1:1687) of the four animals. The other four immunized macaques (NHPs 5 to 8) were inoculated using a repeated low-dose (RLD; 10 TCID50) challenge regimen. In this case, the immunized monkeys (pseudovirus SHIVDH12-V3AD8 titers ranging from 1:1393 to 1:2314; table S1) and control monkeys both rapidly became infected (median times = 5.5 and 5.2 weeks, respectively) (Fig. 6C), indicating no protection as a result of vaccination.
Figure 6. Immunization conferred minimal protection against intrarectal challenge of a heterologous SHIV in NHPs.
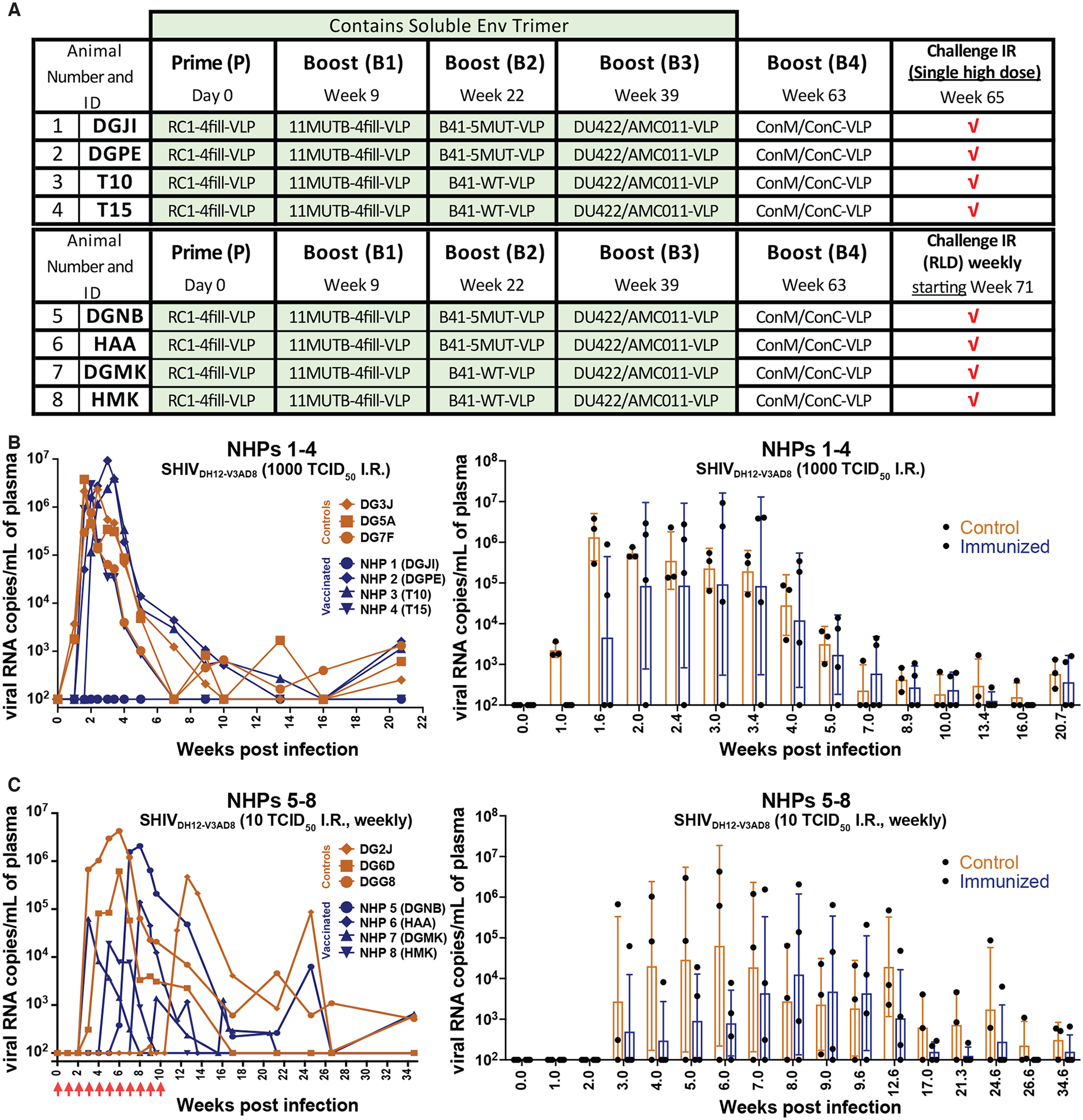
(A) The schedule for prime, boosting, and SHIVDH12-V3AD8 challenge for NHPs is shown, including NHP numbers and IDs. (B) Left: Plasma viral loads are shown for macaques challenged intrarectally with a single high dose (1000 TCID50) of SHIVDH12-V3AD8 two weeks after receiving the final boost. Controls are shown in orange and immunized animals are shown in blue. Right: A comparison of viral loads in control versus immunized NHPs is shown as a function of time after infection presented as geometric means with standard deviations. Values for individual NHPs are shown as dots. (C) Left: Plasma viral loads are shown for macaques challenged intrarectally with repeated low doses (10 TCID50) of SHIVDH12-V3AD8 weekly, beginning two-weeks following the final boost. Controls are shown in orange and immunized animals are shown in blue. Right: A comparison of viral loads in control versus immunized NHPs is shown as a function of time after infection presented as geometric means with standard deviations. Values for individual NHPs are shown as dots. Time points were plotted when all animals were tested.
The results of minimal to no protection in SHIV-challenged animals can be evaluated with reference to in vitro neutralization assay results. When pseudotyped SHIVDH12-V3AD8 was used to monitor neutralization activity in the serum of the immunized macaques at the time of virus challenge, neutralization titers were measured in the 1:1393 to 1:9953 range (table S1). By contrast, against replication-competent virus, the titers were much lower (1:39 to 1:256) (table S1). Similarly, high neutralization titers were observed against pseudotyped SHIVSF162.LS, but not against replication-competent SF162P3 (table S1). Finally, no neutralization activity was detected against the highly pathogenic SHIVAD8EO (64), even in an assay using a pseudovirus readout (table S1). Thus the prime-boost immunization regimen did not achieve protection from SHIV challenge, despite high titers against the heterologous SHIVDH12-V3AD8 in the in vitro pseudovirus assay.
This lack of protection following immunization contrasts with previously-reported results obtained following the administration of bNAbs to macaques prior to high-dose challenges with SHIVDH12-V3AD8 (63). On the day of challenge, two animals that received the V3-glycan patch bNAb 10–1074 (28) generated serum neutralization titers in pseudovirus assays of 1:420 and 1:376, respectively, and both were protected from infection. Similarly, two monkeys that were administered the CD4-binding site bNAb 3BNC117 (26) produced neutralization titers in pseudovirus assays of 1:143 and 1:142, respectively, and also resisted SHIVDH12-V3AD8 infection (63). Although the vaccine regimen used in our study generated much higher polyclonal neutralizing titers against SHIVDH12-V3AD8 (table S1), it failed to prevent infection in all but one of eight NHPs. We also note that serum neutralization titers against BG505 pseudovirus elicited in BG505-vaccinated NHPs that were protected against autologous BG505 SHIV challenge (72, 73) were much lower than the SHIVDH12-V3AD8 titers measured here. However, it should be noted that the autologous neutralizing antibody response to BG505 largely consists of antibodies directed against the glycan hole in the vicinity of gp120 residue 241 (47, 50, 67). Thus, the autologous anti-BG505 neutralizing antibodies, like the administered bNAbs, appear to protect at lower potencies than the more heterogeneous responses (Fig. 5A) produced after the vaccination regimen described here. Achieving protection from heterologous SHIV challenge will likely require higher neutralizing titers, measured using replication-competent virus, than exhibited in bNAb-administered or autologously-vaccinated NHPs.
Discussion
To date, no HIV-1 vaccination protocol designed to elicit neutralizing antibodies has elicited protective serum responses against HIV-1 or SHIV challenge in animals with a polyclonal antibody repertoire. Here we report strategies that elicited consistent, although weakly, heterologous neutralizing serum (all immunized NHPs) and mAbs (from representative NHPs). A main hurdle in the elicitation of bNAbs by immunization in wt animals with polyclonal antibody repertoires is the activation and expansion of distracting antibody responses that target non-conserved epitopes of Env. Immunization experiments in rabbits and NHPs have shown that holes in the Env glycan shield, the base of the trimer, and epitopes exposed upon trimer dissociation into protomers represent distracting, non-neutralizing epitopes elicited by SOSIP Env trimers (47, 50, 67). Moreover, additional interference to the bNAb development process may come from antibody responses to conserved epitopes of Env that show poor potential to become bNAbs (21, 22). The design of Env immunogens and immunization strategies to focus the antibody responses to conserved epitopes of Env while preventing off-target responses is crucial for the success of a vaccine protocol in humans.
In this study, we aimed to focus the antibody responses to conserved epitopes of Env by using a series of engineered SOSIP Env trimers followed by native-like SOSIPs in an attempt to gradually induce the maturation of bNAb precursors as previously reported in knock-in mice (23, 74). To mask potentially immunodominant and distracting regions of Env, we introduced extra potential N-linked glycans to the glycan shields of the immunogens and conjugated them to VLPs. To minimize boosting of antibody responses to HIV-1 strain-specific epitopes, we used a series of multi-clade Env immunogens derived from different HIV-1 strains that we conjugated individually or in combinations to VLPs.
Both nsEMPEM and competition ELISA analyses of antibodies elicited following the prime and subsequent boosts suggested that an epitope at or near V3 glycan-patch, as targeted in our immunogen, were the predominant antibodies observed after a prime and initial boosts in rabbits and NHPs, both representing animals with polyclonal repertoires. Although these results are encouraging, both methods of analysis include caveats related to their interpretation. For nsEMPEM, kinetic and affinity properties of the polyclonal Fabs can influence which Fab-Env complexes are observed. Kinetic and affinity differences could also affect results of competition ELISAs. Additionally, a specific mAb used as competitor against a particular epitope, such as the trimer base, may not block all antibodies in that class, and the competitor mAbs do not cover all possible Env epitopes. With these caveats, our results showed that our immunogen design was successful at primarily recruiting antibodies against the desired region of Env in wt animals despite the many neutralizing and non-neutralizing epitopes on HIV-1 Env, a more difficult endeavor compared with previous studies that used modified animals expressing the relevant germline B cell receptor (23, 74). This is in contrast to structural studies of animals immunized with BG505, which predominantly developed antibodies against non-conserved epitopes and did not consistently elicit antibodies with breadth and potency against heterologous Envs (75). In addition, our results showed slight differences in the orientation of Fabs observed by nsEMPEM after the prime and first boosts that were consistent with accommodating the gp120 N156 glycan (which had been removed in our priming immunogen), consistent with higher-resolution cryo-EM structures of Env trimers bound to prime and boost mouse mAbs and with serum neutralization data demonstrating potent neutralization of both RC1 and 11MUTB pseudoviruses as well as weaker, but relatively broad, neutralization of a diverse panel of HIV-1 strains. Thus our prime-boost strategy elicited antibodies that targeted the desired epitope and exhibited neutralization of heterologous strains.
We previously reported cryo-EM structures of mAbs cloned from primed NHPs and wt mice complexed with RC1, revealing recognition of the V3-glycan patch (38). A recent report suggested that the Env interactions of two of our prime-elicited NHP mAbs, Ab874NHP and Ab897NHP, resembled those of 43A, a non-neutralizing V1/V3 antibody identified from a rabbit immunized with soluble BG505 SOSIP.664 (76), suggesting that antibodies induced by RC1–4fill-VLP could not mature to become neutralizing. However, Table S2 of that paper (76) showed more Ab874NHP and Ab897NHP interactions with residues within the conserved V3-glycan patch GDIR motif and fewer interactions with V1 as compared with 43A, implying that the NHP antibodies elicited after priming with RC1–4fill-VLPs could represent a different class than the non-neutralizing 43A antibody. In addition, Ab874NHP (and Ab1170NHP) showed interactions with the gp120 N332 glycan, another desirable property for a bNAb precursor that targets the V3-glycan patch.
Here we demonstrated that at least a subset of the antibodies induced after a RC1–4fill-VLP prime (38) did indeed mature to become neutralizing, both with respect to polyclonal serum neutralization and as individual mAbs isolated after boosting. Indeed, all three NHPs from which mAbs were isolated produced neutralizing antibodies that showed V3-glycan patch targeting. Although the observed antibody potencies were weak compared with human donor-derived bNAb potencies, the induced responses were heterologously neutralizing, including neutralization of strains representing the global HIV-1 pandemic (71), suggesting that these mAbs recognize a conserved epitope on HIV-1 Env. It is also noteworthy that nsEMPEM showed that the RC1–4fill and subsequent boosting immunogens induced predominant responses that mapped to the Env trimer at the targeted epitope, such as at or near the V3-glycan patch. Since V3-glycan patch bNAbs adopt different poses, a successful V3-glycan patch immunogen need not elicit antibodies that bind the V3 region of Env in the same way as known V3-glycan patch bNAbs. Indeed, our results suggest it is possible to elicit different classes of V3-targeting bNAbs using an RC1-based immunogen, which is potentially a more desirable property in a vaccine than inducing antibodies derived from only one V gene segment lineage.
Although the elicited antibodies were at least partially specific for the V3-glycan patch, nsEMPEM demonstrated that our boosting regimen needs further optimization to minimize off-target responses. In particular, we observed increased antibodies against non-neutralizing epitope(s) at the Env trimer base after repeated boosts and the presence of dissociated Env trimers. Biochemical and biophysical characterization of our immunogen nanoparticles indicated that improved purification methods can be used to ensure the immunization does not include free Env trimer to prevent access to the trimer base. In addition, off-target responses to the Env base might be eliminated by using a modified version of RC1–4fill that conjugates efficiently to SpyCatcher nanoparticles (RC1–3fill, with one fewer introduced PNGS in the vicinity of the glycan hole in BG505). Finally, immunogens could incorporate mutations to prevent trimers from dissociating into protomers that will induce off-target non-neutralizing antibodies, such as SOSIPs with introduced disulfide bonds (DS-SOSIPs) (77, 78).
The analysis of the neutralization activity elicited by our sequential immunization protocol revealed an important caveat in the preclinical methods commonly used to evaluate vaccine candidates. We observed a discrepancy between the serum neutralization activity in assays involving pseudoviruses versus replication-competent viruses, with the activity against the latter being lower. These results are in agreement with previous observations showing higher IC50 values for antibodies against PBMC-derived HIV-1 viruses compared with pseudoviruses produced in HEK cells (72, 79–82) and with the finding that differences in the cells used to produce viruses can affect the virus’ neutralization sensitivities (79).
A recent study demonstrated that comparable or lower pseudovirus antibody titers to those seen in our study provided protective responses in NHPs (72); however, the animals in those studies were challenged with a BG505 SHIV that was autologous to the BG505 immunogen. Comparison of our results with those studies suggests that protection from heterologous HIV-1 or SHIV infection will require higher in vitro neutralization potencies than exhibited by the heterologously neutralizing antibodies elicited in our study. For example, recent clinical trials to evaluate bNAb-mediated protection from HIV-1 infection in humans demonstrated a lack of protection from infection by HIV-1 strains against which the administered bNAb VRC01 exhibited weak in vitro potencies (HVTN 704/HPTN 085 and HVTN 703/HPTN 081). This finding, when considered in combination with our results and with studies of the limited ability of weakly-neutralizing anti-HIV-1 bNAbs to delay viral rebound after cessation of anti-retroviral therapy (83), suggest that high titers of broadly-active serum neutralizing activity will be required for a protective vaccine against HIV-1. As a potential way around an absolute requirement for eliciting highly potent neutralizing antibody activity for protection after vaccination, a recent study evaluating protection from an autologous SHIV challenge in BG505-immunized NHPs showed that low serum neutralizing titers (<1:300 in a pseudovirus assay) were sufficient for protection when anti-Gag T cell responses were simultaneously induced (73). Thus, even weakly neutralizing antibody responses, if sufficiently broad, might be protective if combined with an optimal T cell-inducing vaccination regimen.
As discussed above, our study has several limitations including the use of immunogens that allow off-target antibody elicitation. Thus, other vaccination regimes may further improve neutralization activity. For example, future efforts to use immunogens designed to target two or more epitopes could be beneficial to prevent resistance to antibodies against a single Env epitope, a strategy that should be possible by vaccination since antibodies are elicited against multiple epitopes on HIV-1 Env during natural infection (18, 40, 84–86). Using prime and boost immunogens targeting multiple epitopes that circumvent the problems identified in this study, perhaps in combination with T cell-inducing immunization regimens, our results combined with other studies suggest it may be possible to elicit protective immune responses against HIV-1 infection using a sequential vaccination strategy.
Methods
Study Design
The objective of this study was to investigate whether an immunization regimen with RC1 as a priming immunogen can elicit anti-HIV bNAbs and protect against heterologous SHIV challenge in NHPs. RC1 was selected as a prime based on a previous study (38), which demonstrated RC1 could elicit antibody precursors to the V3 epitope in both wild-type mice and NHPs. NHPs (n=8) were primed and boosted four times and serum isolated after each immunization was tested for neutralization against a panel of HIV and SHIV strains. We used ELISAs and nsEMPEM to map the epitopes of elicited antibodies on HIV-1 Envs and single-cell sorting of antigen-positive memory B cells from immunized macaques identified multiple mAbs that demonstrated breadth and potency against a panel of heterologous HIV strains, including the challenge strain SHIVDH12-V3AD8. Two of these mAbs were then structurally characterized using cryo-EM, which revealed their mode of binding to HIV-1 Env. Following immunization, NHPs were challenged with high-dose rectal challenge (n=4) or repeated low-dose rectal challenge (n=4) and viral loads were assessed over 21 to 35 weeks. The study was not blinded and all data were included except where specified in the text or figure legends. All NHP experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Immunogen expression and purification
Env immunogens were expressed as soluble SOSIP.664 native-like gp140 trimers (43) as described (38). For SpyTagged trimers, a C-terminal SpyTag sequence (13 residues) was added to allow formation of an irreversible isopeptide bond to SpyCatcher protein (49). We also produced AviTagged BG505 and clade B B41 SOSIPS for B cell sorting experiments, and untagged and SpyTagged versions of 11MUTB-4fill (a version of the BG505-related 11MUTB SOSIP (42) containing PNGSs to add glycans to gp120 positions N230, N241, N289, and N344), B41–5MUT (a B41 version of the BG505-related 5MUT SOSIP (42) including a PNGS at gp120 position 289, B41wt (a B41 SOSIP with an introduced PNGS at position 289), clade C Du422, clade B AMC011 with an introduced PNGS at gp120 position 230, consensus C SOSIP (ConC) (56), and a consensus M SOSIP (ConM) (55). Amino acid sequences of SpyTagged SOSIP immunogens are shown in table S5.
Soluble SOSIP Envs were expressed by transient transfection in HEK293–6E cells (National Research Council of Canada) or Expi293 cells (Life Technologies) and purified from transfected cell supernatants by 2G12 or NIH45–46 immunoaffinity chromatography and size exclusion chromatography (SEC) as described (87). Soluble Envs were stored at 4°C in 20 mM Tris pH 8.0, and 150 mM sodium chloride (TBS) (untagged and AviTagged versions) or 20 mM sodium phosphate pH 7.5, 150 mM NaCl (PBS) (SpyTagged versions).
VLP preparation and conjugation
SpyCatcher-AP205 VLPs were expressed in bacteria and purified as described (48, 88). SpyTagged SOSIP immunogens were incubated at a 2 to 3–fold molar excess with SpyCatcher-VLPs for 12 to 24 hours at room temperature in phosphate-buffered saline (PBS). Conjugated VLPs were separated from free Env trimers by SEC on a Superdex 200 (fig. S7D) or Superose 6 (fig. S7E) column. Conjugation of Env trimers was verified by SDS-PAGE (fig. S7C). Immunogen concentrations for immunizations were estimated by comparing to known amounts of the analogous unconjugated Env trimer on a SDS-PAGE gel.
Conjugated VLPs were examined by nsEM (fig. S7A and D). Purified VLPs were diluted to about 10 μg/mL immediately before adding 3 μL to a glow-discharged ultrathin C film on a holey carbon support film, 400 mesh, Cu grid (Ted Pella). After blotting, the grids were stained by uranyl acetate and then imaged using a FEI Tecnai T12 transmission electron microscope operating at 120 keV with a Gatan Ultrascan 2k × 2k CCD camera. Each image was collected using a 1 second exposure at about 2 μm defocus and 42,000× magnification, resulting in 2.5 Å per pixel.
SPR binding studies
SPR experiments were performed using a T200 (Biacore). To determine if the Env trimer base was accessible for antibody binding on a densely-conjugated VLP, we covalently immobilized 12N IgG (47) on a CM5 chip using primary amine chemistry according to manufacturer’s instructions to a coupling density of about 3000 resonance units (RUs). 0.1 mg/mL RC1 SOSIP or 0.1 mg/mL RC1-VLPs (based on SOSIP concentration) were then injected at a flow rate of 30μL/min for 60 seconds, followed by monitoring of the dissociation phase for 300 seconds. Flow cells were regenerated with 10 mM glycine pH 3.0 at a flow rate of 90 μl/min. Sensorgrams were processed and plotted using the Biacore Evaluation Software.
Animal immunizations
Eight rhesus macaques (Macaca mulatta) of Indian genetic origin, two-to-four years of age, were housed in a biosafety level 2 NIAID facility and cared for in accordance with Guide for Care and Use of Laboratory Animals Report number NIH 82–53 (Department of Health and Human Services, Bethesda, 1985). All animal procedures and experiments were performed according to protocols approved by the IACUC of NIAID, NIH. The NHPs used in this study did not express the MHC class I Mamu-A*01, Mamu-B*08 and Mamu-B*17 alleles. NHPs were immunized subcutaneously in the medial inner forelegs and hind legs (total of 4 sites per animal) with approximately 100 μg of the indicated SOSIP-VLPs adjuvated in IscoMPLA (375 U/animal) as described (38). Animals were immunized and blood samples were drawn from naïve and immunized macaques at the time points indicated in Fig. 1A, tables S1 and S2, and Fig. 6. Blood samples were processed to serum or plasma by centrifugation in serum separating tubes or EDTA-treated tubes, respectively. Serum and plasma samples were frozen and stored at −80°C. Lymph node biopsies for single B cell cloning were obtained after Boost 3 and Boost 4 (Fig. 1A).
Four six-month-old New Zealand White rabbits (Covance) were used for immunizations. Rabbits were immunized subcutaneously with ~22 μg of a SOSIP-VLP in an ISCOMs-like saponin adjuvant as described (38, 89). Blood from immunized rabbits was collected from the medial ear artery on weeks 0 and 2 following immunization (test and production bleeds) or the jugular vein/carotid artery after the final boost (terminal exsanguination) by Covance. Serum was processed by Covance laboratory technicians by centrifugation in serum separating tubes and shipped frozen. Procedures in rabbits were approved by the Denver PA IACUC Committee.
In vitro neutralization assays
RC1 and 11MUTB pseudoviruses were generated by transfection of HEK293 cells with a codon-optimized gp160 construct containing RC1 (38) or 11MUTB (42) mutations in gp120 and a wild-type BG505 gp41 as previously described (51, 90). Pseudovirus neutralization assays were conducted using TZM-bl reporter cells as described (51, 90), either in house (table S1) or at the Collaboration for AIDS Vaccine Discovery (CAVD) core neutralization facility (table S2, Fig. 5A). IgG mAbs were evaluated in duplicate with an 8-point, 5-fold dilution series starting at a top concentration of 500 μg/mL. RC1 and 11MUTB pseudovirus assays were repeated at least twice for each value reported in table S1. For polyclonal neutralizations, serum or plasma samples were heat inactivated at 56 °C for 1 hour before being added to the neutralization assays, and then neutralization was evaluated in duplicate with an 8-point, 4-fold dilution series starting at a dilution of 1:20 (heterologous strains) or 1:100 (RC1 and 11MUTB strains). The dilution at which 50% of virus was neutralized (ID50) and the percent of neutralization at a 1:20 dilution (% 1:20) are reported (table S2). The panel of global HIV-1 Env reference clones were obtained through the NIH HIV Reagent Program (ARP-12670) as contributed by Dr. David Montefiori.
For neutralization assays to evaluate NHP serum for neutralization against SHIVs, we used two types of assays (91): (i) a TZM-bl entry assay with a pseudotyped SHIVDH12-V3AD8 and (ii) a TZM-bl infection assay using replication-competent SHIVDH12-V3AD8 virus. The pseudotyped virus expressed the SHIVDH12-V3AD8 Env was conducted as described above. The replication-competent SHIVDH12-V3AD8 virus was made as described (63, 92) by first transfecting HEK293T cells with SHIVDH12-V3AD8 cloned DNA, infecting rhesus PBMCs with the transfected HEK293T cell supernatant, and then collecting the infected rhesus PBMC cell supernatant at 48 hours as the SHIV DH12-V3AD8 replication competent virus stock. For replication-competent virus assays, the protease inhibitor, indinavir (Merck), was added to the medium (final concentration of 1 μM) to prevent a second round of viral replication.
Isolation and purification of rabbit and NHP polyclonal IgG antibodies
Rabbit and NHP polyclonal IgG antibodies were purified from serum samples using 5-mL HiTrap MabSelect SuRe columns (Cytiva). Serum samples were diluted 10-fold with cold PBS, and applied to prepacked columns at 1 mL/min. Bound IgG antibodies were washed with 5 column volumes PBS and eluted with 5 column volumes 0.1M glycine pH 3.0, 100 mM NaCl. Samples were then immediately neutralized with 2M Tris-HCl, pH 8. To produce polyclonal Fab fragments, IgG antibodies were buffer exchanged into PBS and cleaved by papain digestion using activated crystallized papain (Sigma-Aldrich) for 30 to 60 min at 37°C at a 1:100 enzyme:IgG ratio. To remove undigested IgG antibodies and Fc fragments, digested products were applied to a 1 mL HiTrap MabSelect SuRe column (Cytiva) and the flowthrough containing cleaved Fabs was collected. Fabs were further purified by SEC using a Superdex 200 Increase 10/300 column (GE Healthcare Life Sciences) in TBS, before concentrating and storage at 4°C.
nsEMPEM data collection and processing
nsEMPEM methods were adapted from (50, 67). Polyclonal rabbit Fab-Env complexes were formed by incubating 25 μg of a SOSIP.664 trimer with 2–3 mg of polyclonal Fabs in 100 μL total volume either overnight (post-Prime and post-Boost 1) or for 30 minutes (all others) at room temperature. NHP Fab-Env complexes were formed by incubating 12.5 μg of Env SOSIP.664 trimers with 1.25 mg of polyclonal Fabs in 50 μL total volume for 30 minutes at room temperature. Fab-Env complexes were purified by SEC on a Superose 6 increase 10/300 GL column (Cytiva). Fractions containing complexes were pooled and concentrated to 5 to 10 μg/mL before deposition on a freshly glow-discharged 300 mesh carbon-coated copper grid (Ted Pella) and stained with 1.5% (w/v) uranyl formate (Electron Microscopy Sciences).
All Fab-Env complexes were imaged on a 200 kV Talos Arctica transmission electron microscope (Thermo Fisher Scientific) operating at room temperature, equipped with a K3 direct electron detector (Gatan) using SerialEM 3.7 (93), except for the Rabbit 2249 RC1-post-Prime complex, which was imaged on a 120 kV Tecnai T12 equipped with a CCD camera. Images were processed in cryoSPARC v 2.14, and a reference-free particle stack was generated using a Gaussian blob picker (94) or by EMAN2 (95) for the rabbit post-Boost 4 – RC1 dataset. Particles corresponding to Fab-Env complexes were identified by iterative rounds of 2D classification. 2D class averages that displayed structural elements interpreted as Fabs bound to an intact Env trimer or dissociated gp140 protomer were selected for further 2D and 3D classification. Ab initio models generated in cryoSPARC were either reported directly or used for heterogeneous or homogenous refinement. Figures were prepared using UCSF Chimera (96, 97), and Fabs were highlighted in selected 2D classes using IMOD (98).
Isolation of mAbs from immunized NHPs
Single HIV-1 Env-specific B cells were isolated from NHP lymph nodes from lymph nodes (20, 70). Antibody cloning from immunized NHPs and production of the NHP mAbs in Fig. 5 was done as described (20, 70). Additional mAbs were either previously reported (Ab1170NHP, Ab275mur) (38) or isolated from an RC1-primed and sequentially-boosted wt mouse (Ab283mur) using similar methods. V(D)J gene assignments of NHP and murine antibodies were done using IMGT/V-QUEST (99). Sequence alignments were done using Clustal Omega (100).
Epitope mapping by competition ELISAs
Competition ELISAs were performed using a Tecan Evo liquid handling robot. RC1 was randomly biotinylated at primary amines using EZ-link NHS-PEG4 Biotinylation Kit according to the manufacturer’s protocol (Thermo Fisher Scientific), resulting in about 5 biotins per protomer. Randomly biotinylated SOSIP trimers coated on streptavidin plates were verified to exhibit similar properties as counterpart site-specifically immobilized SOSIPs (by a C-terminal tag recognized by an anti-tag antibody) and evaluated for binding to anti-Env antibodies by ELISA. By contrast, SOSIP trimers directly coated onto ELISA plates exhibited aberrant behaviors such as binding to CD4-induced antibodies in the absence of CD4, which was not observed for counterpart experiments using randomly biotinylated or site-specifically immobilized SOSIPs. Biotinylated RC1 (1 μg/mL) was bound overnight at 4°C to a 384-well ELISA plate coated with streptavidin (Thermo Fisher Scientific). The plate was washed with TBS-T, incubated with 10 μg/mL Fab for 2 hours, and then 1:100 dilution of serum, 1 μg/mL IgG (controls in serum competition ELISA), or 10 ug/mL mAb IgG (controls and NHP mAbs; mAb competition ELISA) was added and incubated for 2 hours at room temperature. Bound IgG was detected using an horseradish peroxidase-conjugated anti-human IgG Fc secondary antibody (Southern Biotech Cat # 2014–05; 1 hour, room temperature) and developed with SuperSignal ELISA Femto Substrate (Thermo Fisher Scientific). Relative light units (RLU) were measured and the signal for each Fab-IgG pair was normalized to the signal for the IgG when no blocking Fab was present. Measurements were performed in technical quadruplicates and the data presented are representative of two independent experiments.
Antibody production and purification
IgG mAbs were expressed by transient transfection in Expi293 cells or HEK293–6E cells and purified from cell supernatants using MabSelect SURE (Cytiva) or protein A or G columns (GE Healthcare) as described (38, 61). 6xHis-tagged Fabs were purified by Ni-NTA chromatography (Cytiva) and SEC (61). The Ab283mur mouse Fab for structural studies was obtained by digesting Ab283mur IgG at 1–5 mg ml−1 with ficin (Sigma Aldrich) and purified by protein G (GE Healthcare) and SEC chromatography (101), followed by monoQ 5/50 (GE Healthcare) ion exchange chromatography. The common iGL of the PGT121 and 10–1074 bNAbs (28) was expressed as an IgG.
Single-particle cryo-EM
For the Ab283MUR–RC1–8ANC195 complex structure, purified Ab283mur Fab, 8ANC195 Fab, derived from a bNAb directed against the gp120-gp41 interface (61), and RC1 SOSIP trimer were incubated at a 3:1 molar ratio of Fab per protomer overnight at room temperature. For the Ab1170NHP–RC1–8ANC195 complex structure, purified Ab1170NHP Fab, 8ANC195 Fab, and RC1 SOSIP trimer were incubated at a 3:1 molar ratio of Fab per protomer overnight at room temperature.
The complexes were subjected to SEC on a Superdex 200 column (GE Life Sciences), and 3 μL of purified complex (0.4 mg/mL for Ab1170NHP–RC1–8ANC195 and 1.35 mg/mL for Ab283mur–RC1–8ANC195) was added to freshly glow-discharged 300 mesh, 1.2/1.3 Quantifoil copper grids and then blotted for either 2 or 3.5 seconds with Whatman no. 1 filter paper at 22°C and 100% relative humidity using a Mark IV Vitrobot. Samples were then vitrified in 100% liquid ethane. Micrographs were collected on a Talos Arctica TEM (Thermo Fisher Scientific) operating at 200 kV and processed in Relion (102, 103) for both complexes (table S4).
For the Ab283mur–RC1–8ANC195 complex, automated data collection was carried out using EPU 2 software (Thermo Fisher Scientific) with a Falcon 3EC direct electron detector (Thermo Fisher Scientific). Micrographs were motion-corrected including dose-weighting using MotionCorr2 within Relion (102). The non-dose-weighted micrographs were used for contrast transfer function (CTF) estimation in CTFFind-4.1 (104). After discarding micrographs with poor CTF fits and signs of visible ice, particles were auto-picked, extracted, and 2D classified. Particles from selected 2D classes were used for an initial refinement with an initial map that was created by 60 Å-low-pass-filtering a map created from a model of BG505–10–1074–8ANC195 complex using molmap in UCSF Chimera (96, 105). This model was then used as an input model for 3D classification. Particles from selected 3D classes were refined again into a 3D model, which was calculated using C3 symmetry applied and a mask that did not include Fab CHCL domains. From there, particles were polished and movie-refined before a final 3D refinement that resulted in a map with a gold standard Fourier shell coefficient (FSC) calculation of 5.2 Å.
For the Ab1170NHP–RC1–8ANC195 complex, automated data collection was carried out using SerialEM software (93, 106) with a Gatan K3 camera. Micrographs were motion-corrected including dose-weighting using MotionCorr2 within Relion (102). The non-dose-weighted micrographs were used for CTF estimation in CTFFind-4.1 (104). After discarding micrographs with poor CTF fits and signs of visible ice, particles were auto-picked, extracted, and 2D classified. Particles from selected 2D classes were used for 3D classification with an initial map that was created by 50 Å-low-pass-filtering a map created from a model of the BG505–8ANC195 complex (61) using molmap in UCSF Chimera (96, 105). Selected particles were then 3D refined using C1 symmetry into a map with a gold standard FSC calculation of 4.5 Å. Coordinates for the Ab283mur–RC1 portion of the Ab283mur–RC1–8ANC195 complex were generated by docking chains from reference structure (PDB 6ORQ) into the map using UCSF Chimera (96, 105). The model was then modified to the correct sequence. Subsequently, the model was refined iteratively in Phenix (107, 108) and Coot (109). Structure figures were made using UCSF Chimera (96) or PyMol (110).
NHP challenge experiments
The origin and preparation of the SHIVDH12-V3AD8 stock used for virus challenge in NHPs was previously described (63). In “high dose” virus challenges, NHPs were inoculated intrarectally with 1000 TCID50 of SHIVDH12-V3AD8 2 weeks following Boost 4. For “repeated low dose challenges,” animals were also challenged intrarectally 2 weeks following the Boost 4, but with 10 TCID50 of SHIVDH12-V3AD8 and every week thereafter, until infection was established, as described (91). Viral RNA amounts in plasma were determined by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) (ABI Prism 7900HT sequence detection system; Applied Biosystems) as described (111).
Statistical analyses
All raw, individual level data are shown in data file S1. For the statistical analysis of the mean titers after Boost 2 against the SHIVDH12-V3AD8 pseudovirus in comparisons of the four NHPs that received the B41–5MUT-VLP boost with the four NHPs that received the B41wt-VLP boost, we performed a two-tailed t-test assuming unequal variances using Excel. For both of these groups of NHPs, the Shapiro-Wilk test on their titer measurements does not reject the null hypothesis of normal distribution, so this t-test was applicable. For analyzing the Kaplan-Meier curves indicating the fraction of NHPs (immunized versus control) with viral loads below the limit of detection following challenge, two methods were used: the Log-rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test. Geometric means were calculated for the viral loads presented in Fig. 6B and C since the values varied over many orders of magnitude.
Supplementary Material
Acknowledgements
We thank members of the Bjorkman, Martin, and Nussenzweig laboratories for discussions, M. Howarth (Oxford) for providing plasmids and advice for VLP expression and purification, J. Moore (Weill Cornell Medical College), R.W. Sanders and M.J. van Gils (Amsterdam UMC) for SOSIP expression plasmids, J. Vielmetter, P. Hoffman, and the Protein Expression Center in the Beckman Institute at Caltech for expression assistance, T. Eisenreich and S. Tittley for animal husbandry, and K. Gordon for flow cytometry. Electron microscopy was performed in the Caltech Cryo-EM Center with assistance from S. Chen and A. Malyutin.
Funding:
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) Grant HIVRAD P01 AI100148 (to P.J.B. and M.C.N.), the Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (CAVD) grant INV-002143 (to P.J.B., M.C.N., and M.A.M.), a Bill and Melinda Gates Foundation grant #OPP1146996 (to M.S.S.), the Intramural Research Program of the NIAID (to M.A.M.), NIH P50 AI150464 (to P.J.B.), and NIH grants UM1 AI144462 and P01 AI048240 (to D.J.I.). A.E. was supported by a NIH K99/R00 grant, A.T.D. and M.E.A. were supported by NSF Graduate Research Fellowships, and C.O.B. was supported by the Hanna Gray Fellowship Program from the Howard Hughes Medical Institute and the Postdoctoral Enrichment Program from the Burroughs Wellcome Fund. M.C.N. is an HHMI investigator.
Footnotes
Competing interests: H.B.G., P.J.B., A.E., and M.C.N. are coinventors on a patent (HIV Vaccine Immunogens; PCT/US2019/063619) covering RC1 immunogens. All other authors have no competing interests.
Data and materials availability:
All data associated with this study are in the paper or supplementary materials. nsEMPEM and cryo-EM reconstructions were deposited in the Electron Microscopy Data Bank (EMDB, https://www.emdataresource.org/) and the associated accession numbers are listed in table S3. ISCOMs-like saponin adjuvant is available from Darrell Irvine under a materials transfer agreement with MIT.
Bibliography
- 1.Baba TW et al. , Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat Med 6, 200–206 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Balazs AB et al. , Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 48, 81–88 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichberg JW, Murthy KK, Ward RH, Prince AM, Prevention of HIV infection by passive immunization with HIVIG or CD4-IgG. AIDS Res Hum Retroviruses 8, 1515 (1992). [DOI] [PubMed] [Google Scholar]
- 4.Emini EA et al. , Prevention of HIV-1 infection in chimpanzees by gp120 V3 domain-specific monoclonal antibody. Nature 355, 728–730 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Hessell AJ et al. , Broadly neutralizing monoclonal antibodies 2F5 and 4E10 directed against the human immunodeficiency virus type 1 gp41 membrane-proximal external region protect against mucosal challenge by simian-human immunodeficiency virus SHIVBa-L. J Virol 84, 1302–1313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hessell AJ et al. , Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat Med 15, 951–954 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hessell AJ et al. , Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog 5, e1000433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mascola JR et al. , Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med 6, 207–210 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Mascola JR et al. , Protection of Macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J Virol 73, 4009–4018 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moldt B et al. , Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc Natl Acad Sci U S A 109, 18921–18925 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parren PW et al. , Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virol 75, 8340–8347 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pietzsch J et al. , A mouse model for HIV-1 entry. Proc Natl Acad Sci U S A 109, 15859–15864 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibata R et al. , Neutralizing antibody directed against the HIV-1 envelope glycoprotein can completely block HIV-1/SIV chimeric virus infections of macaque monkeys. Nat Med 5, 204–210 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Bonsignori M et al. , Antibody-virus co-evolution in HIV infection: paths for HIV vaccine development. Immunol Rev 275, 145–160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwong PD, Mascola JR, HIV-1 Vaccines Based on Antibody Identification, B Cell Ontogeny, and Epitope Structure. Immunity 48, 855–871 (2018). [DOI] [PubMed] [Google Scholar]
- 16.McCoy LE, Burton DR, Identification and specificity of broadly neutralizing antibodies against HIV. Immunol Rev 275, 11–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.West AP Jr. et al. , Structural Insights on the Role of Antibodies in HIV-1 Vaccine and Therapy. Cell 156, 633–648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao HX et al. , Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496, 469–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roark RS et al. , Recapitulation of HIV-1 Env-antibody coevolution in macaques leading to neutralization breadth. Science 371, eabd2638 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z et al. , A broadly neutralizing macaque monoclonal antibody against the HIV-1 V3-Glycan patch. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Escolano A, Dosenovic P, Nussenzweig MC, Progress toward active or passive HIV-1 vaccination. J Exp Med 214, 3–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haynes BF, Mascola JR, The quest for an antibody-based HIV vaccine. Immunological Reviews 275, 5–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Escolano A et al. , Sequential Immunization Elicits Broadly Neutralizing Anti-HIV-1 Antibodies in Ig Knockin Mice. Cell 166, 1445–1458 e1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao X et al. , Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: implications for evasion of immune responses and design of vaccine immunogens. Biochem Biophys Res Commun 390, 404–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonsignori M et al. , Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J Virol 85, 9998–10009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheid JF et al. , Sequence and Structural Convergence of Broad and Potent HIV Antibodies That Mimic CD4 Binding. Science 333, 1633–1637 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein F et al. , Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153, 126–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mouquet H et al. , Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci U S A 109, E3268–3277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou T et al. , Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abbott RK et al. , Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 48, 133–146 e136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G, Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol 161, 5373–5381 (1998). [PubMed] [Google Scholar]
- 32.Dosenovic P et al. , Anti-HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc Natl Acad Sci U S A 115, 4743–4748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shih TA, Meffre E, Roederer M, Nussenzweig MC, Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol 3, 570–575 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Tas JM et al. , Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwickert TA et al. , A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med 208, 1243–1252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong R et al. , Antibody Lineages with Vaccine-Induced Antigen-Binding Hotspots Develop Broad HIV Neutralization. Cell 178, 567–584.e519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kong L et al. , Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat Struct Mol Biol 20, 796–803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Escolano A et al. , Immunization expands B cells specific to HIV-1 V3 glycan in mice and macaques. Nature 570, 468–473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker LM et al. , Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freund NT et al. , Coexistence of potent HIV-1 broadly neutralizing antibodies and antibody-sensitive viruses in a viremic controller. Science translational medicine 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sok D et al. , A Prominent Site of Antibody Vulnerability on HIV Envelope Incorporates a Motif Associated with CCR5 Binding and Its Camouflaging Glycans. Immunity 45, 31–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steichen JM et al. , HIV Vaccine Design to Target Germline Precursors of Glycan-Dependent Broadly Neutralizing Antibodies. Immunity 45, 483–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanders RW et al. , A next-generation cleaved, soluble HIV-1 Env Trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog 9, e1003618 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garces F et al. , Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell 159, 69–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gristick HB et al. , Natively glycosylated HIV-1 Env structure reveals new mode for antibody recognition of the CD4-binding site. Nat Struct Mol Biol 23, 906–915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scharf L et al. , Structural basis for germline antibody recognition of HIV-1 immunogens. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy LE et al. , Holes in the Glycan Shield of the Native HIV Envelope Are a Target of Trimer-Elicited Neutralizing Antibodies. Cell reports 16, 2327–2338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brune KD et al. , Plug-and-Display: decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Scientific reports 6, 19234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zakeri B et al. , Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A 109, E690–697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bianchi M et al. , Electron-Microscopy-Based Epitope Mapping Defines Specificities of Polyclonal Antibodies Elicited during HIV-1 BG505 Envelope Trimer Immunization. Immunity 49, 288–300 e288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montefiori DC, Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Current protocols in immunology / edited by John E. Coligan … [et al Chapter 12, Unit 12 11 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Pugach P et al. , A native-like SOSIP.664 trimer based on an HIV-1 subtype B env gene. J Virol 89, 3380–3395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Gils MJ et al. , An HIV-1 antibody from an elite neutralizer implicates the fusion peptide as a site of vulnerability. Nature Microbiology 2, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li M et al. , Genetic and Neutralization Properties of Subtype C Human Immunodeficiency Virus Type 1 Molecular env Clones from Acute and Early Heterosexually Acquired Infections in Southern Africa. Journal of Virology 80, 11776–11790 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rutten L et al. , A Universal Approach to Optimize the Folding and Stability of Prefusion-Closed HIV-1 Envelope Trimers. Cell reports 23, 584–595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sliepen K et al. , Structure and immunogenicity of a stabilized HIV-1 envelope trimer based on a group-M consensus sequence. Nature Communications 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH et al. , A Broadly Neutralizing Antibody Targets the Dynamic HIV Envelope Trimer Apex via a Long, Rigidified, and Anionic beta-Hairpin Structure. Immunity 46, 690–702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker LM et al. , Broad and Potent Neutralizing Antibodies from an African Donor Reveal a New HIV-1 Vaccine Target. Science 326, 285–289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schoofs T et al. , Broad and Potent Neutralizing Antibodies Recognize the Silent Face of the HIV Envelope. Immunity 50, 1513–1529 e1519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong R et al. , Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 352, 828–833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scharf L et al. , Broadly Neutralizing Antibody 8ANC195 Recognizes Closed and Open States of HIV-1 Env. Cell 162, 1379–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JH et al. , Antibodies to a conformational epitope on gp41 neutralize HIV-1 by destabilizing the Env spike. Nat Commun 6, 8167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shingai M et al. , Passive transfer of modest titers of potent and broadly neutralizing anti-HIV monoclonal antibodies block SHIV infection in macaques. J Exp Med 211, 2061–2074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishimura Y et al. , Generation of the pathogenic R5-tropic simian/human immunodeficiency virus SHIVAD8 by serial passaging in rhesus macaques. J Virol 84, 4769–4781 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lorenzi JCC et al. , Neutralizing Activity of Broadly Neutralizing anti-HIV-1 Antibodies against Primary African Isolates. J Virol 95, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cohen YZ et al. , Neutralizing Activity of Broadly Neutralizing Anti-HIV-1 Antibodies against Clade B Clinical Isolates Produced in Peripheral Blood Mononuclear Cells. J Virol 92, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nogal B et al. , Mapping Polyclonal Antibody Responses in Non-human Primates Vaccinated with HIV Env Trimer Subunit Vaccines. Cell reports 30, 3755–3765 e3757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barnes CO et al. , Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 182, 828–842 e816 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turner HL et al. , Disassembly of HIV envelope glycoprotein trimer immunogens is driven by antibodies elicited via immunization. Sci Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Z et al. , Isolation of single HIV-1 Envelope specific B cells and antibody cloning from immunized rhesus macaques. J Immunol Methods 478, 112734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.deCamp A et al. , Global panel of HIV-1 Env reference strains for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol 88, 2489–2507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pauthner MG et al. , Vaccine-Induced Protection from Homologous Tier 2 SHIV Challenge in Nonhuman Primates Depends on Serum-Neutralizing Antibody Titers. Immunity 50, 241–252.e246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arunachalam PS et al. , T cell-inducing vaccine durably prevents mucosal SHIV infection even with lower neutralizing antibody titers. Nature Medicine 26, 932–940 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steichen JM et al. , A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Antanasijevic A et al. , Polyclonal antibody responses to HIV Env immunogens resolved using cryoEM. bioRxiv, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nogal B et al. , HIV envelope trimer-elicited autologous neutralizing antibodies bind a region overlapping the N332 glycan supersite. Science Advances 6, eaba0512 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Joyce MG et al. , Soluble Prefusion Closed DS-SOSIP.664-Env Trimers of Diverse HIV-1 Strains. Cell reports 21, 2992–3002 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Antanasijevic A et al. , Structural and functional evaluation of de novo-designed, two-component nanoparticle carriers for HIV Env trimer immunogens. PLOS Pathogens 16, e1008665 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Provine NM, Cortez V, Chohan V, Overbaugh J, The neutralization sensitivity of viruses representing human immunodeficiency virus type 1 variants of diverse subtypes from early in infection is dependent on producer cell, as well as characteristics of the specific antibody and envelope variant. Virology 427, 25–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muecksch F et al. , Development of potency, breadth and resilience to viral escape mutations in SARS-CoV-2 neutralizing antibodies. Immunity, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wieczorek L et al. , Mitigation of variation observed in a peripheral blood mononuclear cell (PBMC) based HIV-1 neutralization assay by donor cell pooling. Virology 447, 240–248 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Mascola JR et al. , Recommendations for the design and use of standard virus panels to assess neutralizing antibody responses elicited by candidate human immunodeficiency virus type 1 vaccines. J Virol 79, 10103–10107 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trkola A et al. , Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med 11, 615–622 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Bonsignori M et al. , Maturation Pathway from Germline to Broad HIV-1 Neutralizer of a CD4-Mimic Antibody. Cell 165, 449–463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gao F et al. , Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell 158, 481–491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Medina-Ramirez M et al. , Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J Exp Med 214, 2573–2590 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang H et al. , Asymmetric recognition of HIV-1 Envelope trimer by V1V2 loop-targeting antibodies. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tissot AC et al. , Versatile virus-like particle carrier for epitope based vaccines. PLoS One 5, e9809 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lövgren-Bengtsson K, Morein B, in Vaccine Adjuvants: Preparation Methods and Research Protocols, O'Hagan DT, Ed. (Humana, 2000), pp. 239–258. [Google Scholar]
- 90.Montefiori DC, Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol 485, 395–405 (2009). [DOI] [PubMed] [Google Scholar]
- 91.Gautam R et al. , A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature 533, 105–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shingai M et al. , Most rhesus macaques infected with the CCR5-tropic SHIV(AD8) generate cross-reactive antibodies that neutralize multiple HIV-1 strains. Proc Natl Acad Sci U S A 109, 19769–19774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mastronarde DN, Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Tang G et al. , EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Goddard TD, Huang CC, Ferrin TE, Visualizing density maps with UCSF Chimera. J Struct Biol 157, 281–287 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Pettersen EF et al. , UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 98.Kremer JR, Mastronarde DN, McIntosh JR, Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 99.Brochet X, Lefranc MP, Giudicelli V, IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36, W503–508 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sievers F et al. , Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Diskin R, Marcovecchio PM, Bjorkman PJ, Structure of a clade C HIV-1 gp120 bound to CD4 and CD4-induced antibody reveals anti-CD4 polyreactivity. Nat Struct Mol Biol 17, 608–613 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scheres SH, RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goddard TD et al. , UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hagen WJH, Wan W, Briggs JAG, Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J Struct Biol 197, 191–198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Afonine PV et al. , Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Terwilliger TC, Adams PD, Afonine PV, Sobolev OV, A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps. Nat Methods 15, 905–908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schrödinger L. (The PyMOL Molecular Graphics System, 2011).
- 111.Endo Y et al. , Short- and Long-Term Clinical Outcomes in Rhesus Monkeys Inoculated with a Highly Pathogenic Chimeric Simian/Human Immunodeficiency Virus. Journal of Virology 74, 6935–6945 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McCoy LE et al. , Holes in the Glycan Shield of the Native HIV Envelope Are a Target of Trimer-Elicited Neutralizing Antibodies. Cell reports 16, 2327–2338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scheres SH, RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Escolano A et al. , Immunization expands B cells specific to HIV-1 V3 glycan in mice and macaques. Nature 570, 468–473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. nsEMPEM and cryo-EM reconstructions were deposited in the Electron Microscopy Data Bank (EMDB, https://www.emdataresource.org/) and the associated accession numbers are listed in table S3. ISCOMs-like saponin adjuvant is available from Darrell Irvine under a materials transfer agreement with MIT.