

Graphical abstract
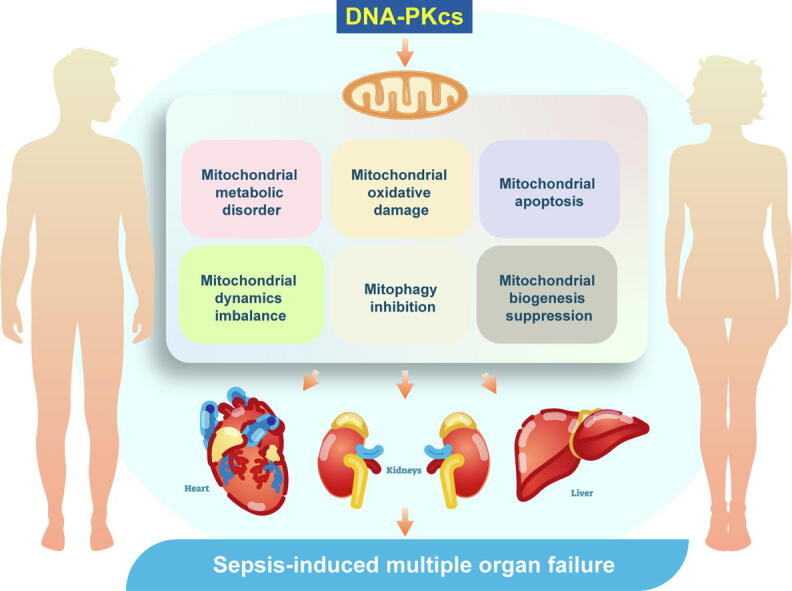
Keywords: DNA-PKcs, MODS, Sepsis, Heart, Mitochondria
Highlights
-
•
DNA-PKcs inhibition attenuates sepsis-related MODS by preserving mitochondrial function and homeostasis.
-
•
Organ-specific deletion of DNA-PKcs sustained myocardial contraction, liver function, and kidney performance in LPS-challenged mice.
-
•
DNA-PKcs deficiency supported cardiomyocyte function through improving mitochondrial respiration.
-
•
DNA-PKcs deficiency alleviated liver dysfunction by inhibiting LPS-induced mitochondrial oxidative stress and apoptosis.
-
•
DNA-PKcs deficiency attenuated kidney dysfunction by normalizing mitochondrial dynamics and biogenesis, as well as mitophagy.
Abstract
Introduction
Multiple organ failure is the commonest cause of death in septic patients.
Objectives
This study was undertaken in an attempt to elucidate the functional importance of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) on mitochondrial dysfunction associated with the development and progression of sepsis-related multiple organ dysfunction syndrome (MODS).
Methods
Cardiomyocyte-specific DNA-PKcs knockout (DNA-PKcsCKO) mice, liver-specific DNA-PKcs knockout (DNA-PKcsLKO) mice, and kidney tubular cell-specific DNA-PKcs knockout (DNA-PKcsTKO) mice were used to generate an LPS-induced sepsis model. Echocardiography, serum biochemistry, and tissue microscopy were used to analyze organ damage and morphological changes induced by sepsis. Mitochondrial function and dynamics were determined by qPCR, western blotting, ELISA, and mt-Keima and immunofluorescence assays following siRNA-mediated DNA-PKCs knockdown in cardiomyocytes, hepatocytes, and kidney tubular cells.
Results
DNA-PKcs deletion attenuated sepsis-mediated myocardial damage through improving mitochondrial metabolism. Loss of DNA-PKcs protected the liver against sepsis through inhibition of mitochondrial oxidative damage and apoptosis. DNA-PKcs deficiency sustained kidney function upon LPS stress through normalization of mitochondrial fission/fusion events, mitophagy, and biogenesis.
Conclusion
We conclude that strategies targeting DNA-PKcs expression or activity may be valuable therapeutic options to prevent or reduce mitochondrial dysfunction and organ damage associated with sepsis-induced MODS.
Introduction
Sepsis is a common cause of critical illness caused by an over-active immune response to bacterial or viral infection [1]. Although the incidence of sepsis is increasing, current therapeutic approaches often show limited success [2]. Optimal care of the septic patients requires early recognition of symptoms and infectious lesions and rapid intervention to mitigate the extent of the immune response. If not treated in a timely manner, septic patients can develop multiple organ dysfunction syndrome (MODS) [3]. MODS is defined as the progressive physiological dysfunction of two or more organ systems where homeostasis cannot be maintained without intervention [4]. Once diagnosed with MODS, the mortality of septic patients can be as high as 28–56%. Therefore, MODS has been regarded as the end stage of a dysregulated systemic inflammatory response. Multiple molecular events, such as endothelial cell dysfunction, oxidative stress, platelet activation, energy metabolism disorder, and uncontrolled cell death have been reported to be involved in MODS [5], [6], [7], [8]. However, the precise mechanisms by which sepsis induces MODS remain to be definitively established. Therefore, there are no available drugs for the treatment of sepsis-induced MODS.
Mitochondria have an indispensable role in regulating cellular metabolism and ATP production through the tricarboxylic acid cycle and the respiratory chain. Besides, many intracellular signals involved in cellular processes such as cell growth, proliferation, differentiation, motility, cell–cell communication, tissue regeneration, and apoptosis, are also influenced by mitochondrial dynamics and function. Our previous studies reported dysregulated mitochondrial homeostasis as a potential mechanism participating in organ dysfunction mediated by myocardial ischemic injury [9], chronic alcoholic liver damage [10], sepsis-related acute kidney injury [11], and hyperglycemia-induced cardiac microvascular dysfunction [12]. Impairment of mitochondrial metabolism occurs early as a consequence of pathogenic insults like those mentioned above. Mitochondrial fission is then typically activated to rapidly augment the number of mitochondria in order to increase ATP production efficiency [13], [14]. However, excessive mitochondrial fission induces mitochondrial oxidative stress and mitochondrial membrane potential reduction, contributing to the leakage of pro-apoptotic proteins from mitochondria to the cytosol [15], [16]. Similar to our findings, previous studies also reported the critical role played by mitochondrial damage in triggering sepsis-related damage in liver, kidney, heart, and vessels [17], [18]. Despite this evidence, the mechanisms underlying mitochondrial dysfunction in the setting of sepsis-related MODS remain incompletely understood.
DNA-dependent protein kinase (DNA-PK) is a serine/threonine protein kinase that plays a crucial role in the cellular DNA damage response. DNA-PK consists of a catalytic subunit (DNA-PKcs) as well as a Ku heterodimer composed of Ku70 and Ku80 subunits[19]. Our previous research unmasked the contribution of DNA-PKcs to the pathogenesis of lipotoxicity-mediated liver damage [20]. We showed that diet-induced nonalcoholic fatty liver disease caused NR4A1-mediated upregulation of DNA-PKcs in hepatocytes, and the ensuing activation of the transcription factor p53 led to aberrant mitochondrial fission through assisting Drp1 translocation from the cytoplasm onto the mitochondrial surface [20]. Subsequently, we reported that in the setting of chronic ethanol-induced liver injury increased expression of DNA-PKcs in hepatic cells represses protective mitophagy, resulting in the accumulation of dysfunctional mitochondria [10]. Consistent with our findings, DNA-PKcs-induced, p53-dependent mitochondrial damage was also identified as a mechanism underlying vascular calcification [21]. Along these lines, deletion of DNA-PKcs was shown to confer neuroprotection against kainate-induced excitotoxic injury, an effect related to abrogation of Bax-induced apoptosis and maintenance of mitochondrial function [22]. Similarly, we recently demonstrated that ischemic cardiomyopathy in mice could be alleviated through specific ablation of DNA-PKcs in cardiomyocytes [23]. We further showed that upon loss of DNA-PKcs, the levels of Bax inhibitor-1 were increased, which prevented the activation of Bax-dependent apoptosis [23]. Collectively, the above findings demonstrate that DNA-PKcs exerts significant control over mitochondrial homeostasis in various cells and organs. Considering the close relationship between DNA-PKcs and inflammation-related organ dysfunction resulting from various pathogenic insults, in this study we explored whether DNA-PKcs contributes to sepsis-related MODS by impairing mitochondrial function and dynamics.
Materials and methods
Mouse models
Cardiac-specific homozygous DNA-PKcs knockout (DNA-PKcsCKO) mice [23], liver-specific DNA-PKcs knockout (DNA-PKcsLKO) mice, and homozygous DNA-PKcsf/f mice, used as the control group, were generated as previously described [10]. KapCre mice (Stock No. 008781) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). DNA-PKcsf/f mice were crossed with KapCre mice to generate kidney tubular cell-specific DNA-PKcs knockout (DNA-PKcsTKO) mice. Genetic background was adjusted by crossing these mice with C57BL/6 inbred mice for more than 5 generations. To establish a sepsis model [24], these mice received LPS (20 mg/kg body weight) for 24-hour. Both sexes of mice were randomly assigned to experimental groups. All animal experiments were performed with randomization and allocation concealment and analyzed in a blind fashion, and were approved by the Chinese PLA General Hospital Institutional Animal Care and Use Committee.
Echocardiographic analysis
The echocardiographic method used in this study has been described previously [25]. Mice were anesthetized using 12 μL/g body weight of 2.5% Avertin (Sigma-Aldrich, USA), and echocardiography was performed using ultrasonography (Visual Sonics Vevo 770, Toronto, Canada) with a 30 MHz linear ultrasound transducer. Two-dimensional guided M−Mode measurements of LV internal diameter were obtained from at least three beats and then averaged. LV end-diastolic diameter (LVEDD), interventricular septal wall thickness (IVSWT), and posterior wall thickness (PWT) were obtained at the time of the apparent maximal LV diastolic dimension, whereas LV end-systolic diameter (LVESD) was obtained at the time of the most anterior systolic excursion of the posterior wall. LV fractional shortening was calculated as follows: fractional shortening=(LVEDD-LVESD)/LVEDD × 100. All the analyses were performed in a blinded manner regarding the genotype of mice.
Cell culture
H9C2 cells (American Type Culture Collection) were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM) supplemented with fetal calf serum (10%), 2 mM of L-glutamine, 100 U/mL of penicillin, and 100 g/mL of streptomycin at 37 °C in 5% CO2. Only cells with a low number of passages (6–10) were used for the experiments. Normal human liver cells (L02), purchased from American Type Culture Collection (Manassas, VA, USA) were cultured in high-glucose DMEM supplemented with fetal calf serum (10%), 2 mM of L-glutamine, 100 U/mL of penicillin and 100 g/mL of streptomycin at 37 °C in 5% CO2 [10]. HK2 cells (American Type Culture Collection) were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM) [26] supplemented with fetal calf serum (10%), 2 mM of L-glutamine, 100 U/mL of penicillin and 100 g/mL of streptomycin at 37 °C in 5% CO2. To induce a sepsis model in vitro, these cell lines were incubated with LPS (10 μg/ml for 24 h) [24].
Western blotting
A total of 30 µg of tissue was minced and lysed in 2% SDS using a glass homogenizer. Cell debris was pelleted at 13,000g for 10 min at 4 °C. Protein concentration of supernatants was estimated using the BCA assay using BSA as a standard. Samples were separated by electrophoresis on 5%-15% gradient Bis–Tris SDS-Gels (Bio-Rad, Hercules, CA, USA) and transferred to PVDF membranes. Protein transfer was controlled by membrane staining with Direct Blue. After blocking, membranes were incubated with primary antibodies. The corresponding IgG HRP-conjugates were revealed using a chemiluminescent substrate (BioRad) and images were scanned by a luminescence image quantifier (ImageQuant LAS 4000, GE Healthcare, Chicago, IL, USA).
Immunohistochemistry
The hearts from DNA-PKcsf/f and/or DNA-PKcsCKO mice subjected to LPS were frozen in optimal cutting temperature compound (Sakura Finetek), sectioned, and stained with primary IL-6 (Abcam, ab208113) and MCP1 (Abcam, ab8101) antibodies at 1/150 dilution overnight at 4 °C. Sections were conjugated with HRP antibody (Vector Laborato ries) at room temperature for 2 h then covered with DAB (Vector Laboratories), nuclei were counterstained in hematoxylin, and slides were mounted with Vectashield mounting medium (Vector Laboratories). Subsequently, all fields were observed under light microscopy (Nikon) at × 10 magnification. Control experiments without primary antibody demonstrated that the signals observed were specific. Positive areas for IL-6 and MCP1 were semi-quantified by an image processing program (ImageJ) according to our previous studies [27], [28].
Mitophagy assay
Mito-Kemia (mt-Kemia) is a mitochondrially localized pH-sensitive ratiometric probe that allows differential imaging of cytoplasmic and lysosomal mitochondrial localization. Determination of mt-Kemia fluorescence ratio in cells has been described in our previous study [25]. Following experimental treatments, we calculated cell areas with a high 561/457 fluorescence ratio, indicating localization of mt-Kemia in lysosomes. For cell counting, cell borders were defined with fluorescence excitation set at 457 nm.
Elisa
Concentrations of mouse BUN (MyBioSource, Inc., MBS751125), Creatinine (Cr, MyBioSource, Inc., MBS2540563), ASpartate Transaminase (AST, MyBioSource, Inc., MBS8800779), ALanine Transaminase (ALT, MyBioSource, Inc., MBS2022063), LDH (MyBioSource, Inc., MBS8801243), CK-MB ELISA kit (BioVision, Milpitas, E4607), Troponin I (MyBioSource, Inc., MBS5316602), Caspase-3 (MyBioSource, Inc., MBS733100), ATP (MyBioSource, Inc., MBS724442), Caspase-9 (LifeSpan BioSciences, LS-F32788), GSH (MyBioSource, Inc., MBS267424), GPX (MyBioSource, Inc., MBS776262), SOD (MyBioSource, Inc., MBS034842), Mitochondrial complex respiration I (complex I, MyBioSource, Inc., MBS912812), Mitochondrial complex respiration II (complex II, MyBioSource, Inc., MBS108909), Mitochondrial complex respiration III (complex III, Abcam, ab287844), Lactic Acid (Creative Diagnostics, DEIA-BJ2418), Bax (Abcam, ab233624) were measured using ELISA kits according to the manufacturer’s instructions.
Mitochondrial membrane potential determination
For TMRE (Tetramethylrhodamine ethyl ester, perchlorate) quantification, cells were incubated in media containing 30 nmol/L TMRE at 37 °C for 10 min. Cells were analyzed using a BioTek fluorescence reader with excitation at 549 nm and emission at 575 nm.
Immunofluorescence
H9C2 cardiomyocytes were seeded in 12-well dishes containing a cover glass. Cells were washed in PBS and fixed with 4% PFA at room temperature for 20 min followed by permeabilization with 0.3% Triton X-100 in PBS for 5 min. The fixed cells were blocked with 5% normal goat or donkey serum in PBS for 1 h at room temperature and incubated with primary antibodies in PBS at 4 °C overnight. Secondary donkey anti-mouse Alexa594 (ab150112, Abcam, UK) or donkey anti-rabbit Alexa488 (ab150061, Abcam) antibodies were used to detect immunoreactivity. Slides incubated with non-immune IgG and secondary antibodies were used as negative controls. Slides were imaged using an upright fluorescence microscope (DS-Ri2, Nikon, Japan). The primary antibodies used in the present study were as follows: MMP9 (Abcam, ab204850, 1:500), VCAM1 (Abcam, ab271899, 1:500), IL6 (Abcam, ab179570, 1:500), TNFα (Abcam, ab1793, 1:1000), TOM20 (Abcam, ab78547, 1:500).
Electron microscopy (EM) and histological analysis
Conventional EM was performed as described previously [28]. Hematoxylin and eosin, Sudan IV, and TER119 staining were imaged using a Zeiss Axioscope 2 microscope and an AxioCamMR3 camera (Carl Zeiss, Germany). Image acquisition was performed with AxioVision40 version 4.6.3.0 software (Carl Zeiss). To determine acute kidney injury, images were taken by an Olympus BX51 using polarized light microscopy. All digitized images were analyzed by Image Pro Plus Software 7.0 (Media Cybernetics). Areas of interest were drawn within Image Pro Plus to measure the acute kidney injury score as previously described [29].
Quantitative PCR
Total RNA was isolated from freshly harvested mouse tissues using a single cell RNA purification kit (51800, Norgen Biotek, Ontario, Canada). We measured RNA yield (58900, Norgen Biotek) and performed reverse-transcriptase reactions (4106228, Bio-Rad) according to the manufacturers’ instructions. Quantitative PCR was performed for 40 cycles on a QuantStudio 6 Flex Real-Time PCR system using TaqMan assays as recommended by the manufacturer, with GAPDH as control gene. We calculated fold difference values of gene expression according to the delta-delta CT method.
Statistical analysis
All statistical analyses were performed with GraphPad Prism 9 (GraphPad Software). Normal distribution of data was assessed using the Shapiro-Wilk test. For normally distributed data comparing two experimental groups, statistical analyses were performed using unpaired two-tailed t-tests. Statistical analyses of normally distributed data from three or more groups were performed using one-way ANOVA with post-hoc Tukey’s multiple comparison tests. To determine the influence of two categorical independent variables on one continuous dependent variable, two-way ANOVA with post-hoc Tukey’s multiple comparison tests were applied. P < 0.05 was used to determine statistical significance. Normally distributed data are presented as mean and standard error of the mean.
Results
DNA-PKcs deletion attenuates sepsis-induced myocardial damage
To understand the role of DNA-PKcs in sepsis-related myocardial damage, we used cardiac specific DNA-PKcs knockout (DNA-PKcsCKO) mice generated as described previously [9]. LPS was administrated into DNA-PKcsf/f mice (control group) and DNA-PKcsCKO mice for 24 h. Echocardiography was used to observe changes in myocardial function. As shown in Fig. 1A-C, left ventricular ejection fraction (LVEF) was reduced in response to LPS treatment, whereas this alteration could be attenuated by DNA-PKcs depletion. In addition, the value of fractional shortening (FS) was impaired by LPS in control but not in DNA-PKcsCKO mice. Similarly, the E/A ratio, a parameter related to myocardial relaxation, was blunted in control mice but preserved upon DNA-PKcs ablation. Serum levels of cardiac damage biomarkers, including LDH, CK-MB, and Troponin T, were also significantly elevated in LPS-treated control mice compared to DNA-PKcsCKO mice (Fig. 1D-F). Next, electron microscopy was used to observe the effect of DNA-PKcs deletion on myocardial ultrastructure in LPS-treated mice. As shown in Fig. 1G, compared to the control (non-LPS) group, LPS treatment induced myocardial swelling and muscle fiber dissolution. However, these alterations were undetectable in LPS-treated DNA-PKcsCKO mice.
Fig. 1.

DNA-PKcs deletion attenuates sepsis-induced myocardial damage. (A-C) Echocardiography was used to analyze myocardial function in control and DNA-PKcsCKO mice administered LPS. (D-F) ELISA was used to analyze the concentrations of LDH, CK-MB, and Troponin T in mouse serum. (G) Electron microscopy was used to assess myocardial ultrastructure. (H-K) Immunohistochemistry was used to detect the expression of IL-6 and MCP1 in myocardium after LPS treatment. (L-N) qPCR was applied to analyze the transcription of IL-6, TNFα, and MCP1 in cardiac tissue. *p < 0.05.
The nature and extent of the inflammatory response have been regarded as key regulators of septic cardiomyopathy. Therefore, we asked whether DNA-PKcs deletion was associated with repression of inflammation in the LPS-exposed myocardium. Immunohistochemistry assays confirmed that the expression of IL6 and MCP1 were induced by LPS in control cardiomyocytes but was instead unaffected after deletion of DNA-PKcs (Fig. 1H-K). Moreover, qPCR analysis of myocardial tissue demonstrated that the levels of IL-6, TNFα, and MCP1 were rapidly increased after exposure to LPS (Fig. 1L-N). However, stimulation of pro-inflammatory cytokine secretion was obviously inhibited in DNA-PKcsCKO mice. These results demonstrated that DNA-PKcs knockdown significantly reduces sepsis-related myocardial damage in mice.
DNA-PKcs deletion normalizes mitochondrial metabolism in LPS-treated cardiomyocytes
To investigate the cardioprotective mechanisms of DNA-PKcs deletion in the setting of myocardial sepsis, we focused on potential changes in mitochondrial function, a main determinant of cardiomyocyte contraction and relaxation. As shown in Fig. 2A, ATP generation in H9C2 cardiomyocytes was significantly repressed by LPS, whereas this alteration was not observed in DNA-PKcs-depleted cells. Moreover, after LPS exposure, glucose levels were increased, whereas the concentration of lactic acid was reduced, in culture media from control, but not DNA-PKcs-depleted, cardiomyocytes (Fig. 2B-C). These results indicated that DNA-PKcs ablation rescues defective glucose metabolism induced by LPS in cardiomyocytes. To further analyze whether mitochondrial energetics in cardiomyocytes are regulated by DNA-PKcs during sepsis, mitochondrial respiratory function was next assessed. As shown in Fig. 2D-E, compared to the control group both mitochondrial state-3 and state-4 respiration rates were blunted by LPS, while these changes were attenuated upon deletion of DNA-PKcs. Based on these findings, ELISA was next used to analyze potential changes in the activity of the mitochondrial respiratory complex. As shown in Fig. 2F-H, compared to the control group, the activities of complexes I-III were reduced upon exposure to LPS and this alteration could be prevented by DNA-PKcs deletion. These results indicated that mitochondrial metabolism and respiration can be sustained by DNA-PKcs deletion in LPS-exposed cardiomyocytes.
Fig. 2.

DNA-PKcs deletion preserves mitochondrial metabolism and respiration in cardiomyocytes exposed to LPS. (A) ATP production was determined by ELISA. (B-C) The concentrations of glucose and lactic acid in the media of cultured cardiomyocytes were measured by ELISA. (D-E) Analysis of mitochondrial respiration. (F-H) ELISA assessment of mitochondrial respiration complex activity. *p < 0.05.
Deletion of DNA-PKcs reduces sepsis-related liver dysfunction
Our previous studies reported the contribution of DNA-PKcs to mitochondrial dysfunction associated with alcohol-related liver damage [10]. In the present study, we used hepatocyte-specific DNA-PKcs knockout (DNA-PKcsLKO) mice to investigate whether DNA-PKcs is also implicated in sepsis-related liver injury through induction of mitochondrial dysfunction. After LPS administration, peripheral blood was collected and serum levels of ALT and AST, and the AST/ALT ratio, were determined by ELISA. Compared to the control group, LPS treatment elevated ALT, AST, and the AST/ALT ratio. However, DNA-PKcs deletion prevented LPS-mediated liver dysfunction through normalization of the concentrations of ALT and AST (Fig. 3A-C). As in septic cardiomyopathy, the inflammatory response is a key factor involved in sepsis-related liver dysfunction. Therefore, immunofluorescence was applied to observe potential changes in MMP9 and VCAM1, two proteins involved in extracellular matrix degradation and leukocyte-endothelial cell adhesion, respectively. As shown in Fig. 3D-F, compared to the control group hepatic expression of both MMP9 and VCAM1 was rapidly increased in response to LPS administration. However, LPS treatment failed to alter the expression of MM9 and VCAM1 in liver tissue from DNA-PKcsLKO mice. Consistent with these findings, the transcription of IL-1, IL-6, and TNFα was primarily enhanced by LPS in the livers of control mice but not in those from DNA-PKcsLKO mice (Fig. 3G-I). These observations indicate that sepsis-related liver dysfunction is greatly attenuated by DNA-PKcs inhibition.
Fig. 3.

Deletion of DNA-PKcs reduces sepsis-related liver dysfunction. (A-C) Serum levels of ALT, AST, and the AST/ALT ratio, were determined by ELISA in control and DNA-PKcsLKO mice administered LPS. (D-F) Immunofluorescence was applied to observe changes in MMP9 and VCAM1 expression in liver tissue. (G-I) qPCR was applied to analyze the transcription of IL-1, IL-6, and TNFα in hepatic tissue. *p < 0.05.
DNA-PKcs depletion inhibits LPS-mediated oxidative stress and apoptosis in hepatocytes
To further assess the hepatoprotective effects of DNA-PKcs deletion during sepsis, we investigated potential alterations in markers of oxidative stress and apoptosis, two factors known to contribute to sepsis-related hepatic failure. Thus, fluorescent detection of mitochondrial and cytoplasmic ROS generation was performed in both control and DNA-PKcs-depleted L02 hepatocytes treated with LPS. As shown in Fig. 4A-C, the levels of both mitochondrial and cytoplasmic ROS in control cells were drastically augmented in response to LPS treatment. In contrast, the production of mitochondrial and cytoplasmic ROS was inhibited in DNA-PKcs-deficient hepatocytes (Fig. 4A-C). Furthermore, we found that after LPS treatment the expression of the intracellular anti-oxidative enzymes GSH, GPX, and SOD was suppressed in control cells, but was instead maintained at near-normal levels after deletion of DNA-PKcs (Fig. 4D-F). These data suggested that silencing of DNA-PKcs preserves cellular antioxidant activities and thus neutralizes excessive ROS production in hepatocytes in the setting of sepsis-mediated liver dysfunction.
Fig. 4.

DNA-PKcs deletion reduces LPS-mediated mitochondrial oxidative stress and apoptosis in hepatocytes. (A-C) Immunofluorescence staining of mitochondrial ROS and cytoplasmic ROS was performed in control and DNA-PKcs-depleted L02 hepatocytes following LPS treatment. (D-F) ELISA was used to analyze changes in SOD, GPX, and GSH expression in cultured liver cells. (G-H) The expression of caspase-3 and caspase-9 was measured in LPS-exposed hepatocytes through ELISA. (I-J) ELISA analysis of Bax expression and mPTP opening. *p < 0.05.
To assess the potential relationship between DNA-PKcs expression and sepsis-induced apoptosis, the expression of caspase-3 and caspase-9, two major apoptotic markers, was measured through ELISA in cultured hepatocytes. As shown in Fig. 4G-H, compared to the control group, the expression of caspase-3 and caspase-9 was stimulated by LPS in control hepatocytes but not in DNA-PKcs-depleted cells. Mitochondria-dependent apoptosis is primarily regulated by mitochondrial permeability transition pore (mPTP) opening and Bax activation. ELISA results showed that following LPS stimulation both Bax expression and mPTP opening were increased in control but not in DNA-PKcs-depleted hepatocytes (Fig. 4I-J). These results suggest that DNA-PKcs silencing inhibits LPS-induced, mitochondrial initiated apoptosis in hepatocytes.
DNA-PKcs deletion sustains kidney function during experimental sepsis
To investigate the action of DNA-PKcs in sepsis-related kidney dysfunction, kidney tubular cell-specific DNA-PKcs knockout (DNA-PKcsTKO) mice were used. Kidney function was first determined by ELISA. As shown in Fig. 5A-B, compared to non-LPS treated mice, serum levels of BUN and creatinine (Cr) were significantly higher in LPS-treated control mice. However, compared to the latter group the concentrations of BUN and Cr were relatively lower in DNA-PKcsTKO mice. H&E staining was next applied to assess renal tubular damage in the experimental groups. As shown in Fig. 5C, the tubular injury index was increased after exposure to LPS in control mice, whereas this phenotypic alteration was not observed in DNA-PKcsTKO mice. Subsequently, immunofluorescence staining of IL-6 and TNFα was conducted to verify whether DNA-PKcs deletion had an anti-inflammatory effect. As shown in Fig. 5D-F, after LPS administration increased expression of IL-6 and TNFα was observed in renal tissue from control mice, and these changes were markedly attenuated in DNA-PKcsTKO mice. In accordance with these findings, qPCR assays showed that the transcription of IL-6 and TNFα was upregulated by LPS exposure in control mice, but downregulated instead upon deletion of DNA-PKcs in tubular cells (Fig. 5G-H). In sum, these data highlight the protective effect of DNA-PKcs silencing in sepsis-mediated kidney dysfunction.
Fig. 5.

DNA-PKcs deletion sustains kidney function in the sepsis setting. (A-B) Serum levels of BUN and creatinine (Cr) were determined by ELISA in LPS-treated control and DNA-PKcsTKO mice. (C) Estimation of the tubular injury index through H&E staining. (D-F) Immunofluorescence analysis of IL-6 and TNFα expression in kidney tissues. (G-H) qPCR was applied to analyze the transcription of IL-6 and TNFα in renal samples. *p < 0.05.
DNA-PKcs deletion sustains mitochondrial homeostasis in LPS-treated renal tubular cells
Our recent studies in a mouse model of acute kidney injury identified abnormal mitochondrial dynamics, defective mitophagy, and impaired mitochondrial biogenesis as key molecular bases of tubular cell damage in response to ischemic stimulus [29], [30]. Therefore, we asked whether DNA-PKcs also impacts mitochondrial genesis and turnover during sepsis-related kidney dysfunction. Immunofluorescence demonstrated that mitochondrial fragmentation was induced by LPS in human renal proximal tubular HK2 cells (Fig. 6A-C). As shown in Fig. 6 D-G, this effect was accompanied by increased levels of the mitochondrial fission-related genes Drp1 and Mff and decreased transcription of the mitochondrial fusion-related genes Mfn2 and Opa1. In contrast, these transcriptional changes were largely attenuated in DNA-PKcs-deficient HK2 cells (Fig. 6D-G). These data suggest that knockdown of DNA-PKcs sustains mitochondrial morphology through normalization of the balance between fission and fusion events in kidney tubular cells exposed to LPS.
Fig. 6.

DNA-PKcs deletion preserves mitochondrial dynamics, biogenesis, and mitophagy in renal cells exposed to LPS. (A-C) Mitochondrial fragmentation was determined in control and DNA-PKcs-deficient HK2 cells through immunofluorescent labeling of TOM20. (D-G) qPCR was applied to analyze the transcription of Drp1, Mff, Mfn2, and Opa1 in HK2 cells. (H-I) Analysis of mitophagy in LPS-exposed HK2 cells using the mt-Kemia assay. (J-K) The transcription of Pgc1α and Tfam in HK2 cells was determined through qPCR. *p < 0.05.
Mitophagy was next assessed in HK2 cells using the mt-Keima reporter assay. Results showed that mitophagy was impaired by LPS in control cells (Fig. 6H-I). In contrast, following treatment with LPS, DNA-PKcs deletion rescued mitophagic activity, as evidenced by an increased number of acidic puncta (Fig. 6H-I). Mitochondrial biogenesis is primarily regulated by transcriptional levels of Pgc1α and Tfam. Results of qPCR assays showed that LPS inhibited Pgc1α and Tfam mRNA expression in control but not in DNA-PKcs-depleted tubular cells (Fig. 6J-K). These results demonstrate that DNA-PKcs silencing maintains homeostatic balance of mitochondrial fission/fusion events and preserves both mitophagy and mitochondrial biogenesis in LPS-challenged kidney tubular cells.
Discussion
MODS is the commonest cause of death in septic patients. Although the pathophysiological mechanisms of MODS have been largely elucidated, its molecular bases remain incompletely characterized and current treatments are often ineffective [31]. Thus, this study was undertaken in an attempt to elucidate the functional involvement of DNA-PKcs in mitochondrial dysfunction in heart, liver, and kidney during development and progression of sepsis-induced MODS. Our results demonstrated that DNA-PKcs inhibition attenuates sepsis-related MODS by preserving mitochondrial function and homeostasis. Through morphometric and histological analyses, we showed that organ-specific deletion of DNA-PKcs significantly sustained myocardial contraction, liver function, and kidney performance in LPS-challenged mice. Molecular assays further illustrated that DNA-PKcs deficiency supported cardiomyocyte function through improving mitochondrial respiration and alleviated liver and kidney dysfunction by inhibiting LPS-induced mitochondrial oxidative stress and apoptosis in hepatocytes and normalizing mitochondrial dynamics and biogenesis, as well as mitophagy, in kidney tubular cells. These results indicate that DNA-PKcs plays a contributing role in sepsis-induced MODS through disturbing key mitochondrial biological processes. Therefore, targeted therapies repressing DNA-PKcs expression or function represent promising strategies for prevention and treatment of sepsis-induced MODS.
A fundamental characteristic of sepsis-induced MODS is extensive systemic inflammation compromising the normal function of cells within different tissues and organs [32]. Sepsis-induced systemic inflammation is accompanied by dysfunctional coagulation, another important feature of MODS, resulting in endothelial and immune cell dysfunction and cellular and molecular alterations, including augmented glycolysis with hyperlactatemia, oxidative stress, endoplasmic reticulum stress, and apoptosis, in affected cells [33], [34], [35], [36]. Based on evidence of impaired ATP production, enhanced oxidative stress, and apoptosis induction, previous studies proposed mitochondrial dysfunction as a potential mechanism underlying sepsis-related MODS [37], [38], [39]. The present study provides further evidence that the mitochondrion is a key target in sepsis. Common features of mitochondrial dysfunction, such as impaired metabolism and respiratory function, redox imbalance, increased fragmentation and decreased biogenesis, as well as repressed mitophagy [27], [40], [41], [42] were detected in mouse heart, liver, and kidney tissues after exposure to LPS. Our molecular analyses identified specific alterations in mitochondrial function and/or dynamics in each organ studied. Decreased ATP supply as a result of LPS-induced mitochondrial metabolic disorder is associated with an inability of cardiomyocytes to contract and relax normally. Increased mitochondrial oxidative stress may trigger activation of the mitochondria-dependent apoptosis pathway, as observed in hepatocytes challenged with LPS. Our findings also show that abnormal mitochondrial dynamics, such as excessive fission and defective fusion, contributes to LPS-mediated mitochondrial damage and dysfunction in kidney tubular cells. Under normal circumstances, mitochondrial quality control mechanisms assure that abnormal mitochondria are recycled by mitophagy and replaced through biogenesis. However, in some pathological settings such as sepsis, these mechanisms fail, resulting in the accumulation of damaged mitochondria, as observed in kidney tubular cells. Thus, and in accordance with previous studies [37], [38], [39], our present data demonstrated that crucial mitochondria-specific processes are disrupted by sepsis. Based on these findings, development of mitochondrial protective strategies represent an important step in order to prevent and treat sepsis-related MODS.
Although mitochondrial dysfunction is a well-established initial signal of sepsis-induced MODS, the resulting alterations in the mitochondrial regulatory network have not yet been studied intensively. The present findings suggest that DNA-PKcs expression promotes mitochondrial damage in heart, liver, and kidney during sepsis-induced MODS, and are thus in accordance with previous studies assessing the role of DNA-PKcs in different pathological conditions. For example, DNA-PK has been reported to promote mitochondrial metabolic decline during aging [43]. Activation of DNA-PKcs through simulated microgravity is associated with apoptosis in human promyelocytic leukemia cells through a mechanism involving ROS overproduction and Bax activation [44]. Notably, defects in DNA-PKcs contribute to symptoms of severe combined immunodeficiency (SCID), and restored expression of the mitochondrial heat shock proteins mtHsp70 and Hsp60 and increased response to anticancer drugs was observed in DNA-PKcs-deficient mouse SCID fibroblasts [45]. These data uncover the close and complex relationship between DNA-PKcs activation and mitochondrial damage.
Several reports addressed the molecular mechanisms by which DNA-PKcs modulates inflammation. For example, upon induction by double-stranded DNA, the absent in melanoma 2 (AIM2) inflammasome interacts with DNA-PKcs to limit its activation, which promotes apoptosis by reducing Akt activation. Accordingly, impaired apoptosis in Aim2−/− cells could be reversed by DNA-PK inhibitors [46]. Another study showed inhibition of genotoxic stress induced by NF-κB, a classical activator of the cellular response to inflammation and DNA damage, in DNA-PKcs deficient cells [47]. Similarly, experiments in an ovalbumin-induced asthma murine model demonstrated a key role for DNA-PK in regulating asthma-induced inflammation, as administration of NU7441, a DNA-PKcs inhibitor, suppressed the inflammatory response in lung airway cells [48]. Meanwhile, in virus infection DNA-PK has been regarded as a DNA sensor for IRF-3-dependent innate immunity [49]. Taken together, these results demonstrate that DNA-PKcs functions as a regulator of inflammation through multiple mechanisms.
There are several limitations in our study. First, since we used only LPS to establish the sepsis-related mouse MODS model in vivo, the effects of DNA-PKcs inhibition on MODS triggered by other sepsis mediators may turn to be different. Besides, the molecular mechanisms by which sepsis would mediate DNA-PKcs activation, as well as those underlying DNA-PKcs-induced mitochondrial damage, were not specifically explored. Further studies should also address whether DNA-PKcs can be detected in the blood and whether DNA-PKcs activity is involved in pathogenic interactions between various organs during sepsis.
Conclusions
In sum, tissue-specific DNA-PKcs ablation markedly attenuates MODS symptoms in heart, liver, and kidney through preventing mitochondrial dysfunction triggered by sepsis. Therapies targeting DNA-PKcs and mitochondrial homeostasis may thus provide substantial benefits for septic patients through alleviating the occurrence of MODS.
Compliance with ethics requirements
All the studies in our experiments are conducted in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). All animal experimental procedures described here were approved by the Animal Care and Use Committees of the Chinese PLA General Hospital (NO. PLA-2020-C00371Z).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
None.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 81900252, 82000537, 82170241, and 81870249). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Peer review under responsibility of Cairo University.
Contributor Information
Hao Zhou, Email: zhouhao@plagh.org.
Xiaoping Fan, Email: fukui-hanson@hotmail.com.
References
- 1.Livingston D.H., Mosenthal A.C., Deitch E.A. Sepsis and multiple organ dysfunction syndrome: a clinical-mechanistic overview. New Horiz. 1995;3(2):257–266. [PubMed] [Google Scholar]
- 2.Rhee C., Klompas M. Sepsis trends: increasing incidence and decreasing mortality, or changing denominator? J Thorac Dis. 2020;12(S1):S89–S100. doi: 10.21037/jtd.2019.12.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramírez M. Multiple organ dysfunction syndrome. Curr Probl Pediatr Adolesc Health Care. 2013;43(10):273–277. doi: 10.1016/j.cppeds.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Ziesmann M.T., Marshall J.C. Multiple Organ Dysfunction: The Defining Syndrome of Sepsis. Surg Infect (Larchmt) 2018;19(2):184–190. doi: 10.1089/sur.2017.298. [DOI] [PubMed] [Google Scholar]
- 5.Gao Y.-L., Zhai J.-H., Chai Y.-F. Recent Advances in the Molecular Mechanisms Underlying Pyroptosis in Sepsis. Mediators Inflamm. 2018;2018:1–7. doi: 10.1155/2018/5823823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fry D.E. Sepsis, systemic inflammatory response, and multiple organ dysfunction: the mystery continues. Am Surg. 2012;78(1):1–8. [PubMed] [Google Scholar]
- 7.Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Curr Opin Crit Care. 2011;17(2):153–159. doi: 10.1097/MCC.0b013e328344b446. [DOI] [PubMed] [Google Scholar]
- 8.Singh S., Evans T.W. Organ dysfunction during sepsis. Intensive Care Med. 2006;32(3):349–360. doi: 10.1007/s00134-005-0038-9. [DOI] [PubMed] [Google Scholar]
- 9.Jin Q., Li R., Hu N., Xin T., Zhu P., Hu S., et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H., Zhu P., Wang J., Toan S., Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Sig Transduct Target Ther. 2019;4(1) doi: 10.1038/s41392-019-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J., Zhu P., Li R., Ren J., Zhang Y., Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020;10(1):384–397. doi: 10.7150/thno.40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H., Wang S., Zhu P., Hu S., Chen Y., Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–346. doi: 10.1016/j.redox.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H., Ren J., Toan S., Mui D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Research Reviews. 2021;66:101250. doi: 10.1016/j.arr.2020.101250. [DOI] [PubMed] [Google Scholar]
- 14.Wang J., Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020;10(10):1866–1879. doi: 10.1016/j.apsb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan Y., Mui D., Toan S., Zhu P., Li R., Zhou H. SERCA Overexpression Improves Mitochondrial Quality Control and Attenuates Cardiac Microvascular Ischemia-Reperfusion Injury. Mol Ther Nucleic Acids. 2020;22:696–707. doi: 10.1016/j.omtn.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J., Toan S., Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: New insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020;156:104771. doi: 10.1016/j.phrs.2020.104771. [DOI] [PubMed] [Google Scholar]
- 17.Donoso-Fuentes A., Arriagada-Santis D. Organ dysfunction syndrome and mitochondrial adaptation in the septic patient. Bol Med Hosp Infant Mex. 2021;78(6):597–611. doi: 10.24875/BMHIM.20000323. [DOI] [PubMed] [Google Scholar]
- 18.Fry D.E. Multiple organ dysfunction syndrome: past, present and future. Surg Infect (Larchmt) 2000;1(3):155–163. doi: 10.1089/109629600750018088. [DOI] [PubMed] [Google Scholar]
- 19.Collis S.J., DeWeese T.L., Jeggo P.A., Parker A.R. The life and death of DNA-PK. Oncogene. 2005;24(6):949–961. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 20.Zhou H., Du W., Li Y.e., Shi C., Hu N., Ma S., et al. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018;64(1):e12450. doi: 10.1111/jpi.12450. https://doi.org/10.1111/jpi.2018.64.issue-110.1111/jpi.12450. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Y.i., Han X.-Q., Sun X.-J., Yang R., Ma W.-Q., Liu N.-F. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. 2020;25(5-6):321–340. doi: 10.1007/s10495-020-01592-7. [DOI] [PubMed] [Google Scholar]
- 22.Neema M., Navarro-Quiroga I., Chechlacz M., Gilliams-Francis K., Liu J., LaMonica K., et al. DNA damage and nonhomologous end joining in excitotoxicity: neuroprotective role of DNA-PKcs in kainic acid-induced seizures. Hippocampus. 2005;15(8):1057–1071. doi: 10.1002/hipo.20123. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H., Toan S., Zhu P., Wang J., Ren J., Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res Cardiol. 2020;115(2):11. doi: 10.1007/s00395-019-0773-7. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y., Jasper H., Toan S., Muid D., Chang X., Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021;45:102049. doi: 10.1016/j.redox.2021.102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H., Zhu P., Wang J., Zhu H., Ren J., Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25(6):1080–1093. doi: 10.1038/s41418-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J., Toan S., Li R., Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem Pharmacol. 2020;174:113832. doi: 10.1016/j.bcp.2020.113832. [DOI] [PubMed] [Google Scholar]
- 27.Li R., Xin T., Li D., Wang C., Zhu H., Zhou H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018;18:229–243. doi: 10.1016/j.redox.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H., Zhu P., Guo J., Hu N., Wang S., Li D., et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J., Zhu P., Li R., Ren J., Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415. doi: 10.1016/j.redox.2019.101415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J., Zhu P., Toan S., Li R., Ren J., Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol Toxicol. 2020;36(4):365–378. doi: 10.1007/s10565-020-09513-9. [DOI] [PubMed] [Google Scholar]
- 31.Polat G., Ugan R.A., Cadirci E., Halici Z. Sepsis and Septic Shock: Current Treatment Strategies and New Approaches. The Eurasian journal of medicine. 2017;49(1):53–58. doi: 10.5152/eurasianjmed.2017.17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall J.C. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29(7 Suppl):S99–S106. doi: 10.1097/00003246-200107001-00032. [DOI] [PubMed] [Google Scholar]
- 33.Baue A.E. Multiple organ failure, multiple organ dysfunction syndrome, and systemic inflammatory response syndrome. Why no magic bullets? Arch Surg. 1997;132(7):703–707. doi: 10.1001/archsurg.1997.01430310017002. [DOI] [PubMed] [Google Scholar]
- 34.Guo J., Tao W.u., Tang D., Zhang J. Th17/regulatory T cell imbalance in sepsis patients with multiple organ dysfunction syndrome: attenuated by high-volume hemofiltration. Int J Artif Organs. 2017;40(11):607–614. doi: 10.5301/ijao.5000625. [DOI] [PubMed] [Google Scholar]
- 35.Shen H.-H., Huang S.-Y., Cheng P.-Y., Chu Y.-J., Chen S.-Y., Lam K.-K., et al. Involvement of HSP70 and HO-1 in the protective effects of raloxifene on multiple organ dysfunction syndrome by endotoxemia in ovariectomized rats. Menopause. 2017;24(8):959–969. doi: 10.1097/GME.0000000000000864. [DOI] [PubMed] [Google Scholar]
- 36.Murray M.J. Sepsis: clinical dilemmas. Yale J Biol Med. 1998;71(6):485–491. [PMC free article] [PubMed] [Google Scholar]
- 37.Dare A.J., Phillips A.R.J., Hickey A.J.R., Mittal A., Loveday B., Thompson N., et al. A systematic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radic Biol Med. 2009;47(11):1517–1525. doi: 10.1016/j.freeradbiomed.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 38.Aswani A., Manson J., Itagaki K., Chiazza F., Collino M., Wupeng W.L., Chan T.K., Wong W.S.F., Hauser C.J., Thiemermann C., Brohi K. Scavenging Circulating Mitochondrial DNA as a Potential Therapeutic Option for Multiple Organ Dysfunction in Trauma Hemorrhage. Front Immunol. 2018;9:891. doi: 10.3389/fimmu.2018.00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parikh S.M., Yang Y., He L., Tang C., Zhan M., Dong Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol. 2015;35(1):108–119. doi: 10.1016/j.semnephrol.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi C., Cai Y., Li Y., Li Y.e., Hu N., Ma S., et al. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018;14:59–71. doi: 10.1016/j.redox.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu H., Jin Q., Li Y., Ma Q., Wang J., Li D., et al. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones. 2018;23(1):101–113. doi: 10.1007/s12192-017-0827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J., Toan S., Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;23(3):299–314. doi: 10.1007/s10456-020-09720-2. [DOI] [PubMed] [Google Scholar]
- 43.Park S.-J., Gavrilova O., Brown A.L., Soto J.E., Bremner S., Kim J., et al. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017;25(5):1135–1146.e7. doi: 10.1016/j.cmet.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh R., Rajput M., Singh R.P. Simulated microgravity triggers DNA damage and mitochondria-mediated apoptosis through ROS generation in human promyelocytic leukemic cells. Mitochondrion. 2021;61:114–124. doi: 10.1016/j.mito.2021.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Um J.H., Kang C.D., Hwang B.W., Ha M.Y., Hur J.G., Kim D.W., et al. Involvement of DNA-dependent protein kinase in regulation of the mitochondrial heat shock proteins. Leuk Res. 2003;27(6):509–516. doi: 10.1016/s0145-2126(02)00264-3. [DOI] [PubMed] [Google Scholar]
- 46.Wilson J.E., Petrucelli A.S., Chen L., Koblansky A.A., Truax A.D., Oyama Y., et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med. 2015;21(8):906–913. doi: 10.1038/nm.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medunjanin S., Putzier M., Nöthen T., Weinert S., Kähne T., Luani B., et al. DNA-PK: gatekeeper for IKKγ/NEMO nucleocytoplasmic shuttling in genotoxic stress-induced NF-kappaB activation. Cell Mol Life Sci. 2020;77(20):4133–4142. doi: 10.1007/s00018-019-03411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Lin J., Shu J., Li H., Ren Z. Oxidative damage and DNA damage in lungs of an ovalbumin-induced asthmatic murine model. J Thorac Dis. 2018;10(8):4819–4830. doi: 10.21037/jtd.2018.07.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferguson B.J., Mansur D.S., Peters N.E., Ren H., Smith G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife. 2012;1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]