

Abstract
Clear cell renal cell carcinoma (ccRCC), the most common type of kidney cancer, is largely incurable in the metastatic setting. ccRCC is characterized by excessive lipid accumulation that protects cells from stress and promotes tumor growth, suggesting that the underlying regulators of lipid storage could represent potential therapeutic targets. Here, we evaluated the regulatory roles of GPR1 and CMKLR1, two G-protein coupled receptors of the pro-tumorigenic adipokine chemerin that is involved in ccRCC lipid metabolism. Both genetic and pharmacological suppression of either receptor suppressed lipid formation and induced multiple forms of cell death, including apoptosis, ferroptosis and autophagy, significantly impeding ccRCC growth in cell lines and patient derived xenograft (PDX) models. Comprehensive lipidomic and transcriptomic profiling of receptor competent and depleted cells revealed overlapping and unique signaling of the receptors granting control over triglyceride synthesis, ceramide production, and fatty acid saturation and class production. Mechanistically, the receptors both enforced suppression of the triglyceride lipase ATGL but also demonstrated distinct functions, such as the unique ability of CMKLR1 to control lipid uptake through regulation of SREBP1c and the CD36 scavenger receptor. Treating PDX models with the CMKLR1-targeting small molecule -NETA led to a dramatic reduction of tumor growth, lipid storage, and clear cell morphology. Together, these findings provide mechanistic insight into lipid regulation in ccRCC and identify a targetable axis at the core of the histological definition of this tumor that could be exploited therapeutically.
Keywords: clear cell renal cell carcinoma, ccRCC, chemerin, GPR1, CMKLR1, ferroptosis, ATGL, lipidomics, hypoxia, HIF
Introduction
Kidney cancer is the 8th most common cancer diagnosed each year in the United States, and clear cell renal cell carcinoma (ccRCC) is the most prevalent subtype accounting for roughly 75% of cases (1,2). Standard of care for localized ccRCC relies heavily on surgical resection (3). However, about 30% of patients develop metastases leading to a five-year survival rate that has traditionally been lower than 10% (4,5). The main molecular alteration in ccRCC is mutation of the von Hippel-Lindau (VHL) gene, resulting in stabilization of the hypoxia inducible factor (HIF) (6) and activation of downstream pathways involved in glycolysis, angiogenesis, and metastasis (7).
The distinct pathological feature of ccRCC that gives the “clear cell” name is the vast accumulation of cytoplasmic lipids and glycogen into lipid droplets in tumor cells, which positively correlates with tumorigenesis (8). Yet, the mechanism and consequences of increased lipid deposition in ccRCC remain incompletely understood. Recent work from our group and others revealed that lipid storage protects tumor cells from endoplasmic reticulum and reactive oxygen species (ROS) stress and promotes tumor growth (9–11). Furthermore, the lipid species stored in the droplets may be involved in signaling cascades that induce tumor metastasis (12), suggesting that lipid metabolism could be a promising therapeutic strategy for renal cancer.
In a previous study, we discovered that patients with ccRCC produce high levels of the adipokine chemerin, which is essential for tumor growth and lipid storage (13). Chemerin is a multifunctional circulating ligand that impacts lipid metabolism, immune regulation, and angiogenesis (14,15). We defined a critical role for chemerin in ccRCC by shielding tumors from lipid-ROS-induced ferroptosis. However, the mechanism by which chemerin promotes its pleiotropic effects in ccRCC remains unknown. Suppressing global chemerin signaling in genetically engineered mice has been shown to lead to metabolic complications such as glucose intolerance (16), raising the possibility of negative side effects of therapeutic targeting chemerin itself. Therefore, deciphering the downstream signaling pathways might identify additional targets with higher specificity.
In order to define signaling mechanisms driven by chemerin, we sought here to identify the functional receptor(s) in ccRCC. The known receptors of chemerin include G-protein coupled receptors (GPCRs) CMKLR1 and GPR1, and the non-signaling CCRL2 (17–19) for which no function has been established. We focused on CMKLR1 and GPR1 and evaluated their roles by genetic and pharmacological manipulation in ccRCC cell lines and PDX models. We found that both receptors are necessary to promote tumor growth and have overlapping contributions in controlling lipid metabolism. However, while both GPR1 and CMKLR1 reduce adipogenic triglyceride lipase and elevate lipid oxidation and ferroptotic death, CMKLR1 uniquely regulates SREBP-mediated de novo lipogenesis, and GPR1 uniquely suppresses autophagy. Using a CMKLR1 small molecule inhibitor, -NETA, we found that CMKLR1 inhibition suppresses tumor growth. Thus, ccRCC relies on extracellular chemerin signaling via previously unknown receptor-specific phenotypes to suppress lipid metabolism, which is essential for cell survival and proliferation, representing a potentially targetable Achilles’ heel.
Materials & Methods
Cell culture.
UOK101, HEK293 and RAW264.5 cells were cultured in DMEM with 10% FBS; 769-P were cultured in RPMI with 10% FBS; and HK-2 were cultured in keratinocyte-SFM growth media supplemented with human recombinant epidermal growth factor (rEGF) and bovine pituitary extract (BPE) as instructed. Cells were cultured at 37°C and 5% CO2. 769-P, HEK293T and HK-2 were obtained from ATCC. UOK-101 was a gift from Dr. W. Marston Linehan (National Cancer Institute, Bethesda). Cells were used for experiments within 15 passages. All cell lines were tested for mycoplasma using a Lonza MycoAlert® PLUS Kit. -NETA was purchased from Cayman Chemical (#12125) and dissolved in PBS.
Animal studies.
All animal experiments were performed in compliance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of the University of Miami Miller School of Medicine. 6-week old female athymic nude mice and SCID mice were used for cell line and patient derived xenograft studies, respectively. Animals were housed socially (3–5 mice per cage) on a 12-hour light/dark cycle in ventilated cages and with access to food and water. The facility had temperature and humidity control and was equipped with HEPA-filtered air. For tumor growth assays, cells were pelleted and resuspended in PBS. 5×106 cells in 100ul solution were injected subcutaneously into each flank of the nude mice. Once palpable tumors were established, a digital caliper was used to measure tumor size twice a week. Tumor volume was calculated using V=1/2(length x width2). For patient derived xenograft studies, subcutaneous tumor implant was performed by making incisions on the left flank of the animals. Tissue chunks with approximately 0.5 g of weight were implanted into the subcutaneous space of the animals. Tumor volume was measured with a digital caliper and calculated with the same formula. For orthotopic tumor assay, tissue chunks were implanted under the capsules of the left kidney. Tumors were palpable and animals’ behaviors were closely monitored for indications to terminate the assay.
RNA isolation and Real Time Quantitative Polymerase Chain Reaction.
Total RNA was extracted using TRIzol (invitrogen) as instructed. The concentration and quality of RNA were determined using Nanodrop 1000 (Thermo). 500 ng of RNA was used to generate cDNA using Quantabio qScript cDNA supermix (Quantabio) according to the manufacturer’s instructions. The cDNA was diluted 20-fold before qRT-PCR gene expression analysis. qRT-PCR was performed using Power SYBR Green PCR master mix (Applied Biosystems) and SYBR green primers (listed in Supplementary Table 1). The Ct values of target genes was normalized to the housekeeping control, -actin. The fold changes were then calculated using the method.
Cloning & Lentiviral transduction.
For the CRISPR knockout model, knockout cell lines were generated by cloning gRNAs (Supplementary Table 1) into the lentiviral vector lentiCRISPRv2 (Addgene), followed by the lentivirus production by HEK293T cell lines. Briefly, HEK293T cells were transfected with a mixture of CRISPR plasmids, PPAX2 and PMD2G using Lipofectamine 3000 transfection reagent (Invitrogen). Empty lentiCRISPRv2 plasmid was used as a control. The media containing the viral particles was used to infect the ccRCC cells in the presence of 10 μg/ml of polybrene after filtering through 0.45 PVDF sterile filter (Santa Cruz).
Protein Extraction and Western blotting.
The cells were washed with 2x ice-cold PBS before they were lysed in RIPS lysis buffer (50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.5% NP-40; 50 mM NaF) for 30 minutes and scraped from plate. The RIPA buffer contains 1/10 1x protease inhibitor tablet (Thermo Scientific) and phosphatase inhibitor tablet (Thermo Scientific). The lysates were centrifuged at 4°C for 20 minutes at 14,000 rpm. The protein lysates were quantified using Pierce BCA protein Assay Kit (Thermo) according to manufacturer’s instructions. The absorbance was measured using Accuris MR-9600 SmartReader (Benchmark Scientific). Between 30 and 50μg of protein samples were mixed with 6x SDS Laemmli loading buffer and boiled at 95°C for 5 minutes. Proteins were run using 4–20% SDS-PAGE gels (Bio-Rad) and transformed onto PVDF membrane (Millipore). The membranes were blocked with 5% non-fat dry milk in TBS-T at room temperature for 1 hour. The membranes were then blotted with primary antibodies diluted in 5% non-fat dry milk in TBS-T overnight. The membranes were washed with TBS-T for 3×5 minutes at room temperature and blotted in secondary antibodies at room temperature for 1 hour. The antibodies used were listed in Supplementary Table 1.
Cell proliferation assays.
Cells were plated at a density of 30,000 cells in triplicate in a 12-well plate with appropriate culture media. Cells were trypsinized and counted using Cell Counter Countess II (Life Technologies) at day 3, 6, 9, and 12.
Clonogenic survival assays.
The cells were plated in triplicate in 6 cm plates (500 cells/well) with appropriate media to measure survival by colony forming unit (CFU). The colonies were counted after 7 and 10 days respectively for 769-P and UOK101, and the experiment was repeated twice.
Cell viability assays.
After treatment with -NETA, 769-P, UOK101, HK-2 and RAW264.5 cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylterazolium bromide (MP Biomedical) to assess cell survival. Briefly, the cells were plated at a density of 1000 cells/plate in 96-well plates and treated with serial-diluted -NETA for 2 days. MTT solution was diluted 5-fold in DMEM media and incubated in 37°C incubator for 4 hours. The media was carefully removed, and the precipitate was dissolved in isopropanol with 0.1% NP-40 and 4 mM HCl. After 30 minutes of incubation at 37°C, the solution was immediately quantified at 560 nm using Accuris MR-9600 SmartReader (Benchmark Scientific).
BODIPY Lipid Droplet Imaging.
Cells were plated on round glass coverslips of 6-well plates. The medium was removed, and cells were washed with room temperature PBS, fixed with 4% formaldehyde for 30 minutes and incubated with 300 μL of 1 μM BODIPY 493/503 (Life Technologies, Cat# D3922) for 15 minutes, protected from direct light. 1 μg/mL of DAPI (Millipore) was added for nucleus staining. The images were acquired using Olympus FV1000 confocal scanning microscope and analyzed using Fiji software (ImageJ).
Oil Red O (ORO) Lipid Droplet Staining.
For tissue section staining, the tissues were frozen in OCT solution (Fisher Healthcare). Embedded tissue blocks were sectioned at 10 μm thickness and stored at −80°C. Before ORO staining, the frozen sections were air dried and fixed in 10% neutral buffered formalin (Sigma-Aldrich, Cat# HT501128). After fixing in 100% propylene glycol (VWR, Cat# 0575) twice, sections were stained with freshly made ORO solution (Sigma-Aldrich, Cat# O0625) followed by staining for hematoxylin. Then, the slides were washed with distilled water and mounted using glycerin jelly (Sigma-Aldrich). The sections were visualized using a VS120 Virtual Slide Microscope scanner (Olympus). For cell staining, the cells plated in 6-well plates in triplicate were rinsed with PBS twice and fixed with 10% formaldehyde at room temperature for 30 minutes. They were then rinsed with 60% isopropanol for 5 minutes, and stained and visualized as above.
EdU incorporation assay.
Click-iT EdU Cell Proliferation Kit (Thermofisher) was used for this assay. Cells were seeded at 0.8×106 in 6 cm dishes and were incubated with 10 μM EdU solution for 4 hours at 37°C, 5% CO2. After incubation, cells were fixed, permeabilized and stained following the manufacturer’s protocol. The samples were then processed by LSR II cytometer instrument (BD 767 Biosciences) and analyzed using FACS DIVA 8.1 software (BD Biosciences).
Lipid Reactive Oxygen Species.
Cells were plated in 6 cm dishes. 10 μM BODIPY-581/591 C11 (Invitrogen, Cat# D3861) was added to the media and incubated for 1 hour at 37°C, 5% CO2. Cells were then trypsinized and washed twice with HBSS. The samples were then processed by LSR II cytometer instrument (BD 767 Biosciences) and analyzed using FACS DIVA 8.1 software (BD Biosciences). The readings for total fluorescence were used as the measurement of the amount of the lipid reactive oxygen species.
Flow Cytometry.
Cells were plated in 6 cm dishes. Upon desired confluency, cells were trypsinized and washed with FACS buffer (PBS with 1% BSA and 0.1% sodium azide). Cells were then suspended in FACS buffer with primary antibody at 1:100 at room temperature for 4 hours. Cells were then washed with FACS buffer and incubated with Alexa Fluor 555 goat anti rabbit secondary antibody 1:1000 at room temperature for 1 hour, washed, and then analyzed.
Immunofluorescence.
Cells were plated in 6-well plates in triplicate on coverslips were rinsed with PBS twice and fixed with 10% formaldehyde at room temperature for 30 minutes. Cells were then blocked with 10% bovine serum in PBS for 1 hour at room temperature and incubated overnight at 4°C with the primary antibody at 1:1000. Then, cells were washed three times with PBS for 5 min each and incubated with Alexa Fluor 555 goat anti rabbit secondary antibody 1:1000 at room temperature. The slides were washed and mounted with DAPI (Abcam). The sections were visualized using a VS120 Virtual Slide Microscope scanner (Olympus).
Oxygen Consumption Rate measurements.
Oxidation consumption rates were measured using Seahorse XF Pro (Agilent) with its compatible Cell Mito Stress Test Kit (Agilent) as instructed. Briefly, cells were plated at 25,000/well density in the provided 96-well plates. Before the assay, cells were washed with PBS and put in sodium-bicarbonate free media with no serum at 37°C for 1 hour. The provided cartridge was then put onto the plates and loaded into the machine. The analysis was performed using Wave Pro (Agilent).
RNA-sequencing.
Total RNA was extracted using TRIzol (Invitrogen) as instructed. All RNA samples were assessed for quantity and purity, and sequenced by Novogene. Analysis was performed on the Novosmart platform.
Untargeted lipidomics and metabolomics.
Samples were analyzed in the Metabolomics Core Facility at the MD Anderson Comprehensive Cancer Center. Two million cells were harvested and washed with ice cold 0.85% ammonium bicarbonate in deionized water. Cells were then scraped down and pelleted at 400g for 3 min. After removing the supernatant, cells were snap-frozen in liquid nitrogen. 200 μL of extraction solution containing 2% Avanti SPLASH LIPIDOMIX Mass Spec Standard and 1% 10 mM butylated hydroxytoluene in ethanol was added for lipid extraction, and run on a Thermo Fisher Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer with a heated electrospray ionization source operated in data dependent acquisition mode, in both positive and negative ionization modes, with scan ranges of 150−827 and 825−1500m/z. An Orbitrap Resolution Of 120,000 (fwhm) was used for MS1 acquisition and spray voltages of 3,600 and−2,900 V were used for positive and negative ionization modes, respectively. Lipid Search Version 5.0 was used for data analysis and processing.
Quantification and Statistical Analysis.
Statistical analyses were performed with Prism software (GraphPad 8). Statistical significance was evaluated with two-tailed unpaired Student’s and Mann-Whitney t tests for comparing two groups. p < 0.05 was considered to represent a statistically significant difference (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significance).
Data Availability.
All raw data are available upon request from the corresponding author. Lipidomic data are available at https://zenodo.org/records/10913324. RNAseq data generated in this study are publicly available at the EMBL ArrayExpress database with accession number E-MTAB-14016 (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-14016). Publicly available data analyzed in this study were obtained from the KIRC dataset of TCGA using GEPIA2 at http://gepia2.cancer-pku.cn/.
Results
GPR1 and CMKLR1 modulate lipid metabolism and are critical for ccRCC growth.
To gain insight into the functional roles of GPR1 and CMKLR1 in ccRCC, and their importance in mediating adipokine signaling, we used CRISPR/Cas9 to knock out the receptors separately in VHL-deficient, clear cell tumor cell lines (769-P and UOK101). After verification of the knockouts (Figures 1A and 1B), we observed that depletion of either protein significantly reduced cell proliferation (Figures 1C and 1D) and colony formation in vitro (Figures 1E and 1F, Supplementary Figures 1A and 1B). Loss of the receptors led to reductions in cell cycle progression indicated by decreased cyclin E expression (Figures 1A and 1B) and decreased BrdU incorporation (Figures 1G, 1H, 1I). Further, we found induction of various cell death mechanisms including apoptosis from elevated cleaved caspase-3 levels (Figures 1A and 1B), and ferroptosis from elevated C11 BODIPY staining indicating increased lipid peroxidation (Figures 1J and 1K). Thus, both GPR1 and CMKLR1 protect ccRCC cell lines from ferroptosis and other forms of death, and are required for cell proliferation in culture conditions.
Figure 1. GPR1 and CMKLR1 are critical for ccRCC growth.

A/B: Western blot analysis showing GPR1 or CMKLR1 knockout and relevant changes in cell growth.
C/D: In vitro proliferation assay of ccRCC cells with GPR1 or CMKRL1 knockout.
E/F: In vitro colony formation of ccRCC cells with GPR1 or CMKRL1 knockout.
G: EdU incorporation flow diagram of ccRCC cells with GPR1 or CMKRL1 knockout.
H/I: Quantification of EdU incorporation.
J/K: Quantification of C11 Bodipy staining.
To probe further into the mechanism, we performed targeted gene expression profiling and observed decreased expression of genes involved in lipid deposition such as cytoplasmic fatty acid binding protein FABP7, lipid droplet membrane proteins PLIN4 and PLIN2, lipoprotein-forming protein APOE, the catalytic enzyme of sphingomyelin hydrolysis to ceramide and phosphocholine SMPD3, and RORC, a rhythmic regulator of genes involved in lipid metabolism (Figures 2A and 2B). Conversely, we observed increased expression of the rate-limiting enzyme for lipid oxidation, CPT1A. To validate the changes at the protein level, we probed for both CPT1A and ADFP, encoded by PLIN2 gene, and observed changes agreeing with the RNA readouts (Supplementary Figures 1C and 1D). Together, the data suggested that losing either receptor induces a shift from lipid deposition to lipid oxidation. We then assessed the impact on lipid deposition specifically by staining with the lipophilic dye Oil Red-O as well as by treating cells with BODIPY (493/503), a dye that emits fluorescence after binding neutral lipids. With both approaches, we found significant reductions in lipid droplets with both genes in both cell lines (Figures 2C and 2D; and Supplementary Figures 1E–1H). We hypothesized that reduced lipid deposition and increased lipid oxidation should result in increased oxygen consumption and performed Seahorse mito-stress analyses. We found that losing either receptor increased both basal and maximal oxygen consumption rates compared to control cells (Figures 2E and 2F, and 2G and 2H). At the same time, we also observed decreased extracellular acidification rates (ECAR), which is indicative of decreased glycolysis (Supplementary Figures 2A and 2B). This suggests that when GPR1 or CMKRL1 is knocked out, cells utilize less glucose for energy production. Additionally, cells pre-treated with the CPT1A inhibitor, Etomoxir, exhibited significantly decreased oxygen consumption that is comparable to the OCR of control cells, attributing the elevation of mitochondrial oxidation as a result of GPR1/CMKLR1 knockout it largely to lipid oxidation (Supplementary Figures 2C and 2D). Overall, the data support the conclusion that both receptors are required to maintain lipid deposition over lethal lipid oxidation in ccRCC.
Figure 2. GPR1 and CMKLR1 regulate lipid metabolism in ccRCC cells.

A/C: qRT-PCR of lipid metabolism-related genes due to GPR1/CMKLR1 knockout.
B/D: Quantification of ORO staining of GPR1/CMKLR1 knockout cell.
E/F: Mito-stress assay of GPR1/CMKLR1 knockout cell.
G/H: Quantification of oxygen consumption rate of ccRCC cells with GPR1 or CMKRL1 knockout.
Lastly, to validate that the reduced fitness of ccRCC cells in the absence of either GPR1 or CMKLR1 results in reduced tumor growth in vivo, we performed xenograft tumor growth assays in nude mice. Critically, knockout cells displayed no decrement in viability measured by trypan blue at the time of inoculation. As expected, we observed dramatic reductions in tumor growth in both cell lines; indeed no tumor growth was observed in the 769-P line (Figures 3A), while only small tumors formed in the UOK101 injected animals (Figures 3B and 3C). Leveraging the small UOK101 tumors, we assessed gene expression (Figure 3D) and lipid deposition (Figures 3E–3G) and found that our results agreed with the in vitro findings for both receptor knockouts. Together, the data demonstrate that both GPR1 and CMKLR1 contribute to the suppression of lipid oxidation and are essential for maintaining lipid metabolic balance thereby supporting tumor growth, results that are reminiscent of those for chemerin (13).
Figure 3. GPR1 and CMKRL1 modulate ccRCC growth and metabolic shift in vivo.
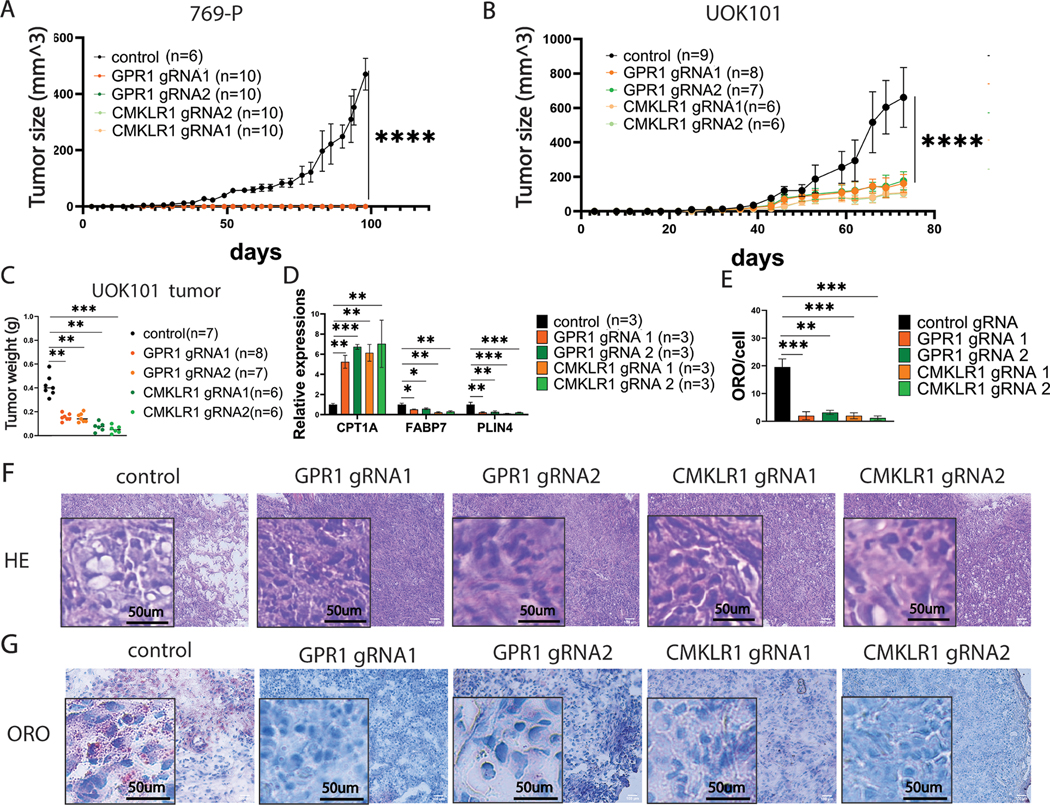
A/B: In vivo tumor assay of ccRCC cells with GPR1 and CMKRL1 knockout.
C: tumor weight of UOK101 with GPR1 and CMKRL1 knockout at terminal point
D: qRT-PCR of key lipid metabolism-related genes of RNAs extracted from UOK101 tumor
E: ORO quantification of UOK101 tumor with receptor knockouts.
F: Hematoxylin and eosin staining of UOK101 tumors.
G: ORO staining of UOK101 tumors.
CMKLR1 and GPR1 have largely similar regulatory roles on the lipidome and transcriptome.
We next sought to understand the extent of lipidome control by the two GPCRs and performed untargeted lipidomic analysis on cells with either GPR1, CMKLR1 or RARRES2 (gene encoding chemerin) CRISPR knockout. Principle component analysis revealed that while all three knockouts exhibited distinct lipid profiles compared to control cells, the CMKLR1 knockout was more closely related to the RARRES2 knockout than GPR1 (Figures 4A and 4B). By Venn diagram analysis of the classes of lipids altered in the three derivatives, it is clear that there are both significant overlaps, but also unique aspects of lipid metabolism controlled by each receptor, which together comprise roughly 75% (308/408 lipids) of the lipidome downstream of chemerin (Supplementary Figures 3A–3D and Supplementary Table 2). Notably, we also determined that expression of each of the three proteins (chemerin, GPR1 or CMKRL1) is not dependent upon one another (Supplementary Figures 3E–3G).
Figure 4. Multiomic analysis revealing the global impact of GPR1 and CMKRL1 on ccRCC.

A: Global heatmap of differentially regulated lipids in 769-P cells.
B: PCA analysis of samples for lipidomic analysis.
C: Enrichment plot of unsaturated lipid species in GPR1/CMKLR1/RARRES2 knockout compared to the control.
D: Bar graph showing ceramide lipid species enrichment in four sample groups.
E: Bar graph showing Phosphatidylethanolamine (PE) lipid species enrichment in four sample groups.
F: Western blot of LC3B in 769-P cells with GPR1 knockout.
G: Western blot of LC3B in 769-P cells with CMKLR1 knockout.
H: Western blot of LC3B in 769-P cells with RARRES2 knockout.
I: Venn diagram showing the number of differentially regulated genes in RARRES2 KO, GPR1 KO and CMKLR1 KO from RNA-seq.
To investigate the meaning of the altered lipids we performed enrichment analysis of the lipid species and observed first that in all three knockout groups there was an overall decreased number of triglycerides that explained the decreased amount of lipid deposition in ccRCC cells (Supplementary Figure 3H). We observed an increased quantities of long chain and polyunsaturated fatty acids (C:20-C:22) that are linked to the lipid oxidative stress associated with ferroptosis (Figure 4C), an expected increase in sphingosine precursors of ceramides known to induce apoptosis (20) as well as a host of inflammatory ceramides themselves in all three groups compared to the control indicative of apoptosis (Figure 4D; Supplementary Figure 3I). We also observed an overall elevation of mitochondrial lipids (Supplementary Figure 4A), suggesting the presence of mitochondrial lipid oxidation, in agreement with in vitro findings.
Aside from the lipid species that confirmed our in vitro findings, we noticed an elevation in PE family fatty acids (Figure 4E; Supplementary Figure 4B), especially in the GPR1 and RARRES2 knockout cohorts, which are suggestive of the induction of autophagy. This led us to investigate if autophagy is involved in chemerin signaling, and uniquely regulated by GPR1, but not CMKLR1. Indeed, western blots confirmed that GPR1 knockout and RARRES2 knockout lead to elevated LC-I to LC-II transition via PE lipidation, a marker for the induction of autophagy; while no change was observed in the CMKLR1 knockout cells (Figures 4F–4H; Supplementary Figures 4C–4E). To further confirm the results, we used Rampamycin to induce autophagy, and chloroquine as an inhibitor to rescue the cells. We performed gene expression analysis on autophagy-related genes including ATG1, GABARAPL1 and SQSTM1 (21) (Supplementary Figures 5A and 5B), and probed again for the LC3–1 to LC3-II transition (Supplementary Figures 5C and Supplementary 6A-F). Indeed, we found that GPR1 and chemerin uniquely control induction of autophagy, which is rescuable with chloroquine. Lastly, we used immunofluorescence staining of LC3 (Supplementary Figures 6E and 6F) to visualize the localization to autophagosomes to finalize the conclusion that, in agreement with the global observation of unique lipid biology controlled by the two receptors, chemerin signaling protects ccRCC from autophagy exclusively through the GPR1 signaling axis.
Next, we performed RNA-seq analysis on cells with either CMKLR1, GPR1, or RARRES2 knockout to understand how the three proteins affect ccRCC cells transcriptionally. Like the lipidomics, a Venn diagram demonstrated overlapping sets of commonly regulated genes between chemerin, GPR1 and CMKLR1; while each receptor also regulates a unique panel of genes separate from each other and from chemerin (Figure 4I, and Supplementary Table 3). Despite the unique genes, CMRKL1 and GPR1 cohorts cover roughly 80% of the genes regulated by chemerin, indicating that these two receptors are indeed the main signal transducers for chemerin. GO enrichment analysis showed similar pathways being affected by the two receptors, including oxidative phosphorylation, lipid metabolism, and electron transport chain (Supplementary Figures 7A-7C). Together, the multiomic datasets alongside phenotypic observations show that GPR1 and CMKLR1 are both important to maintain the ccRCC lipidome.
GPR1 and CMKLR1 inhibit lipolysis by suppressing ATGL, but CMKLR1 uniquely controls lipid uptake.
To understand how the lipid droplets are rewired after ablating receptor signaling, we examined the differentially expressed genes associated with lipid storage versus breakdown. Lipolysis, the process of releasing glycerol and nonesterified fatty acids from triacylglycerol is known to be tightly regulated by lipase family proteins including Hormone Sensitive Ligase (HSL) and Adipose Triglyceride Lipase (ATGL) (22). ATGL, but not HSL, was induced when either receptor was disrupted, an effect that was corroborated by western blot (Supplementary Figure 8A; and Figures 5A and 5B). Likewise, knockout of chemerin, also led to the upregulation of ATGL (Supplementary Figure 8B). To determine if the induction of ATGL was responsible for the depletion of lipid droplets and subsequent oxidation and cell death, we used the ATGL-specific inhibitor Atglistatin and asked whether ATGL inhibition could rescue ccRCC cells. We found that, while not affecting the control cells, Atglistatin treatment at 2 μM for 48 hours reduced apoptosis by measurement of cleaved caspase 3 (Figures 5C and 5D), significantly restored lipid droplet formation measured by ORO (Figures 5E and 5F; Supplementary Figure 8C) and reduced lipid oxidative stress by C11 BODIPY staining (Figures 5G and 5H). Unexpectedly, however, ATGL inhibition did not restore cell proliferation measured by EdU incorporation (Supplementary Figures 9A-9C).
Figure 5. Lipolysis is commonly regulated by GPR1 and CMKLR1 via suppression of ATGL, while CMKLR1 regulates SREBP1c-mediated lipid uptake.

A/B: Western blot of 769-P/UOK101 with GPR1 or CMKLR1 knockout.
C/D: Western blot of 769-P/UOK101 rescued with 2 μM of Atglistatin.
E/F: ORO quantification of 769-P/UOK101 with or without GPR1/CMKLR1 knockout treated with Atglistatin.
G/H: C11 BODIPY quantification of 769-P/UOK101 with or without GPR1/CMKLR1 knockout treated with Atglistatin.
I: ORO quantification of knockout cells treated with external fatty acids.
J: BODIPY (493/503) quantification of knockout cells treated with external fatty acids.
K/L: Western blot of CD36 in ccRCC cells with GPR1 or CMKLR1 knockout.
M/N: Western blot of SREBP1c in ccRCC cells with GPR1 or CMKLR1 knockout.
Mechanistically, ATGL is known to be a target gene of signaling (23). Therefore, we asked if GPR1 or CMKRL1 knockout led to elevation of this signaling pathway. We used qRT-PCR to screen for target genes ADIPOQ and LEP. Indeed, knockout of GPR1 or CMKRL1 led to the elevation of pathway (Supplementary Figure 9D). We then sought to understand the upstream control of the pathway. One of the negative regulators for is DEC1 (24), a HIF target gene (25). Having previously shown that loss of chemerin signaling transcriptionally decreased levels, we asked if knocking out either of the receptors exerted the same HIF suppressive effect and therefore decreased DEC1 levels and elevated signaling. Indeed, from our qRT-PCR results, either GPR1 or CMKRL1 knockout transcriptionally decreased and DEC1 (Supplementary Figures 9C and 9D), supporting elevated signaling, which would expectedly lead to elevated ATGL.
ccRCC cells are also known to actively uptake external fatty acids to enhance their lipid droplets, especially when their lipid deposition pool is depleted (11). Therefore, we asked if external lipid uptake functions as another mechanism downstream GPR1 and CMKLR1 signaling. Interestingly, we observed that when cells were treated with an external fatty acid mixture of oleic acid and linoleic acid (30 μM), only CMKLR1 knockout cells demonstrated decreased lipid uptake compared to controls, shown by both Oil Red-O and BODIPY (493/503) staining (Figures 5I and 5J; and Supplementary Figures 10A-10F). This suggested that pathways uniquely downstream of CMKLR1 may regulate external fatty acid transport. CD36 is a well-established scavenger receptor responsible for translocating fatty acids from the extracellular space to the cytoplasm (26). We therefore asked if CD36 was downstream of CMKLR1. Indeed, we found that CD36 is significantly suppressed both at mRNA and protein levels in CMKLR1 knockout cells, but not GPR1 knockouts (Figures 5K and 5L; Supplementary Figure 11A). Likewise, CD36 is depleted from the cell surface upon CMKLR1 knockout (Supplementary Figure 11B). Knowing that Sterol Regulatory Element-Binding Protein 1c (SREBP1c) is one of the major upstream regulators of CD36, we hypothesized that CMKLR1 pathway disruption leads to reduced SREBP1c signaling, and subsequently reduced CD36-mediated lipid uptake. We first investigated the RNA-Seq data focusing on additional SREBP-1c target genes including FASN, LDLR, LXRa and ABCA1. We found that these genes were suppressed after CMKLR1 knockout and validated the effects by independent qRT-PCR (Supplementary Figures 11C and 11D). We next looked at the protein level of SREBP1c to assess activation by the cleavage to the smaller active form and found that CMKLR1 knockout, but not GPR1 knockout, led to decreased amounts of both cleaved and total SREBP1c protein, thereby suppressing its signaling (Figures 5M and 5N). Together, the data suggest that CMKLR1 uniquely promotes fatty acid uptake, while both pathways are essential to suppress lipolysis and sustain lipid deposition in ccRCC.
Pharmacological inhibition of CMKLR1 using small molecule antagonist -NETA suppresses ccRCC growth.
Having established the criticality of GPR1 and CMKLR1 to control lipid metabolism in ccRCC, we sought pharmacological ways to target the two receptors. Interestingly, from the KIRC dataset of TCGA, while chemerin is significantly overexpressed in tumors versus normal kidney samples (13), only CMKLR1 exhibits a similar behavior (Supplementary Figure 12A), and neither receptor associated with a difference in survival. As a means to antagonize chemerin signaling, however, the genetic data suggests either target could be functionally useful. While there are no readily available agents to suppress GPR1, -NETA is a small molecule antagonist for CMKLR1 that was identified in a screen for inhibitors of the chemerin-stimulated -arrestin2 association with CMKLR1 in an experimental autoimmune encephalomyelitis model of multiple sclerosis (27). It functions by recruiting -arrestin to the carboxyl-terminus of the receptor and results in the internalization of CMKRL1 and termination of signaling (27). We exposed three different ccRCC cell lines as well as three immortalized normal cell lines, including immortalized renal epithelial cells (HK-2), primary renal proximal tubule epithelial cells (PTEC), and RAW-264 macrophages in vitro, and assayed cytotoxicity with both MTT and clonogenic assay (Supplementary Figures 12B and 12C). While the IC50s for the three ccRCC cells (769-P, UOK101 and A498) were 10μM, 15μM and 26μM respectively, the non-transformed cells, HK-2, RAW264 and PTEC were more resistant to -NETA, with IC50 values at 65 μM, 75 μM and 210 μM respectively. Strikingly, there were were even more significant decrements in clonogenic capacity of both ccRCC lines at both 1 and 10 μM doses, where little to no toxicity is observed in the non-tumor lines (Supplementary Figure 12D). We further examined the treated ccRCC cells and observed that like CMKLR1 knockout, -NETA treatment induced ferroptosis (Supplementary Figure 12E). Molecularly, -NETA induced the upregulation of ATGL and suppression of SREBP1c signaling (Figures 6A–6C; Supplementary Figure 12F). Gene expression analyses of CPT1A and FABP7 further suggested that -NETA induced the same metabolic shift from lipid deposition toward lipid oxidation, and changes in lipid metabolism were confirmed by Oil Red-O staining (Figures 6D–6F).
Figure 6. CMKLR1-targeting -NETA induces lipid oxidation and induces a shift in lipid metabolism.

A/B: Western blot probing for ATGL and precursor/active SREBP1c on ccRCC cells treated with -NETA.
C: qRT-PCR of key CMKLR1 regulatory genes on ccRCC cells treated with -NETA.
D: Oil Red-O staining of ccRCC cells treated with -NETA.
E/F: ORO quantification of ccRCC cells treated with -NETA.
To determine the efficacy of -NETA to control tumor growth, we performed xenografts studies with UOK101 cells and dosed the animals with intraperitoneal injection of -NETA at 10 mg/kg three times every week. We found that -NETA was sufficient to significantly reduce tumor growth (Figures 7A and 7B) and decrease the amount of lipid deposition in the tumors that formed visually (Supplementary Figure 12G), and by quantification of Oil Red-O (Figure 7C; Supplementary Figure 12H). Gene expression analysis using tumor RNA validated the genetic studies of CMKLR1 disruption (Supplementary Figure13A).
Figure 7. -NETA suppresses ccRCC growth in both cell line and patient derived xenograft models.

A: UOK101 tumor growth curve with or without the treatment of -NETA.
B: UOK101 tumor weight at the terminal point of -NETA treatment.
C: Normalized ORO staining quantification of UOK101 tumor treated with -NETA.
D: Human ccRCC PDX (XP296) tumor subcutaneous growth curve with or without the treatment of -NETA and the tumor weight at the terminal point
E: XP296 orthotopic tumor weight at the terminal point with or without -NETA.
F: Normalized ORO staining quantification of XP296 subcutaneous tumor treated with -NETA.
G: Oil Red-O staining of XP296 orthotopic tumor with or without the treatment of -NETA.
H: ORO staining quantification of XP296 tumor treated with -NETA.
I: Pathway diagram showing the regulatory role of both GPR1 and CMKRL1 on lipid metabolism in ccRCC.
We next extended the studies to a PDX model that better recapitulates patient tumors. The PDX tissues were provided by Dr. James Brugarolas from UT Southwestern. UTSW-XP296 was generated from the primary tumor of a 47-year-old male that presented with stage IV disease (28). The tumor was locally invasive (pT3a), of clear cell histology, and grade 4, with sarcomatoid features (Supplementary Figures 13B and 13C). The tumor had a frameshift mutation in the VHL gene. In both subcutaneous implantation and orthotopic implantation models, -NETA was again able to suppress tumor growth significantly (Figures 7D and 7E). Critically, we observed no changes to animal health or well-being in any of the treated animals, including no statistically significant differences in body weight at terminal point in subcutaneous model(Supplementary 13B).There was also a striking morphological change in the tumors after CMKLR1 blockade (Supplementary Figures 13C and 13D), that aligned with the loss of ORO staining (Figures 7F-7H; Supplementary Figure 13E), and gene expression of CKLRL1 lipid regulatory genes (Supplementary Figures 13F and 13G). Together, the data suggest CMKLR1 targeting has potential to limit tumor growth in patients by regulating critical lipid metabolic pathways (Figure 7I).
Discussion:
The complexity of the roles of lipids in normal cells, as essential components of membranes, signaling ligands, or energy storage, underlies their altered metabolism as a fundamental hallmark of cancer (29,30). Regulatory networks of lipid metabolism are often harnessed by tumors to meet their demands. ccRCC is histologically known to have a distinct lipid metabolic profile that creates an adipocyte-like storge phenotype (8,31), leading to the concept that ccRCC tumors could potentially harness adipokine signaling to promote aggressive growth. Our group has previously identified a pro-tumorigenic adipokine, chemerin, that is highly expressed by ccRCC tumors and presents a therapeutic vulnerability (13). In the current work, we define the regulatory network elicited by chemerin to control ccRCC lipid metabolism. Our findings indicate that GPR1 and CMKLR1 are two distinct and essential signaling components driving lipid rewiring. Both GPCRs critically support suppression of lipolysis and expansion of the lipid droplets; while CMKLR1 uniquely regulates fatty acid uptake and GPR1 suppresses autophagy. Identifying receptor-mediated cellular dependencies allows for targeted intervention, and we have shown that -NETA can pharmacologically suppress ccRCC growth. Thus, exploiting tumor-lipid alterations may have promise for clinical impact.
Fatty acid oxidation is regarded as a primary bioenergetic source in many tumors, and in particular the rate limiting enzyme of fatty acid transport into the mitochondria, CPT1A, has been linked cell cycle progression in ovarian cancer (32,33). We have previously shown that ccRCC requires suppression of CPT1A and fatty oxidation in order to survive (13) (11). In this study, we determined that blocking the chemerin receptors induces alterations that suppress lipid deposition-related pathways while elevating oxidation-related pathways. Phenotypically, transcriptomic and lipidomic analyses confirm loss of lipid deposition in favor of elevated mitochondrial oxidation, rescuable using CPT1A inhibitor, is the toxic pathway that chemerin signaling necessarily suppresses. Indeed lipid storage rather than consumption is thought to confer a protective buffer to toxic oxidants and may explain why ccRCC excessively produce and update free fatty acids only to apparently set them aside in the lipid droplet (10).
GPR1 and CMKLR1 are the two known functional receptors for chemerin (34). Despite being predicted to have functional homology (35,36), GPR1 and CMKLR1 are clearly not entirely redundant in their regulatory roles in ccRCC based on multiomics and functional analyses. From untargeted lipidomics, although either receptor knockout led to elevation in the amounts of inflammatory and mitochondrial lipid species, which associate with apoptosis and ferroptosis, respectively, inhibition of the chemerin-GPR1 signaling axis led to elevated amounts of PE family fatty acid species and the induction of autophagy that were not as present in the CMKLR1 knockout cells. Pathway enrichment analysis from RNA-seq revealed further differences, for example that CMKLR1 signaling plays a role in macrophage recruitment and complement pathways while GPR1 is involved in kidney development. The observation for CMKLR1 agrees with studies on chemerin as an immune modulator involved in macrophages and dendritic cells (17,37). We have also observed differential regulatory role at the metabolic level. CMKLR1 axis uniquely activated SREBP1c signaling and controls lipid uptake in ccRCC cells. SREBP1c is known to be activated under low cholesterol conditions (38) to increase lipid uptake and synthesis and incorporate into storage (39). It has become increasingly acknowledged that certain fatty acid species can exert regulatory roles on metabolic gene expression to maintain homeostasis (40). Whether the differences in the fatty acid species we observed from the lipidomic analysis contribute to the differential pathways and phenotypes observed in our experiments remains further investigation.
Despite the differences, however, together the two receptors appear to control roughly 75% or more of both lipidomic and transcriptomic effects of chemerin, suggesting they are the operative chemerin signal transducers. At the same time, however, chemerin independent effects may also be evident. CMKLR1 in particular controls five times as many genes as chemerin. The functional overlap would point to chemerin as the effective ligand, but it remains possible other signals also use the same receptors for other means. For example, FAM19A1 has been shown to bind to GPR1 to regulate proliferation and differentiation in neural stem cells (41). A comprehensive understanding of how these two receptors exert overlapping yet differential regulatory roles will need to be further elucidated. Additionally, ATGL, a critical enzyme that initiates the hydrolysis of triglycerides to release fatty acids (42), is a suppressed target of both GPR1 and CMKLR1. The freed up fatty acids resulting from lipolysis fuel the mitochondria for energy production, leading to the accumulation of toxic oxidized lipids and ferroptosis. Indeed, ATGL inhibition decreased elevated lipid peroxidation and rescued cell death. This suggests that suppression of ATGL is critical in protecting ccRCC from lipid-related death and is a crucial target for chemerin signaling. However, ATGL inhibition did not restore all chemerin related phenotypes (e.g. proliferation), confirming that additional endpoints of chemerin signaling exist downstream of the two receptors, such as the known effects on AKT and ERK signaling (19).
-NETA, 2-(-naphthoyl) ethyltrimethylammonium iodide, is a small molecule CMKLR1 antagonist (27) that has been used to treat multiple sclerosis in pre-clinical models. Its ability to penetrate blood-brain barrier has made it an appealing molecule to treat chemerin/CMKLR1 induced neuroinflammatory diseases such as preeclampsia and tumors such as neuroblastoma (43,44), and led to the development of second generation compounds (45). Limited studies have been conducted on using -NETA as a therapeutic agent to treat solid tumors other than glioblastoma and ovarian cancer (46,47). We have demonstrated that -NETA can be effectively used to suppress ccRCC growth through modulation of lipid metabolism with no noted side effects, in agreement with other reports (45). Analysis of harvested tumor tissue showed the expected pathways are affected. This suggests that -NETA is an on-target CMKLR1-specific antagonist, and that its derivatives could potentially be used for clinical purposes to treat ccRCC. As an additional potential benefit, chemerin-CMKLR1 is known to cause recruitment on inflammatory macrophages which may be immune suppressive. Indeed, ccRCC is generally described as an immune privileged microenvironment due to the high content of myeloid derived suppressor cells (48). Thus how -NETA would affect tumor associated macrophages or other myeloid derivatives in ccRCC is unknown, but may lead to enhancement of immunotherapy. Application of -NETA or other chemerin inhibiting approached to a syngeneic model of ccRCC would allow answers to these critical questions.
In summary, we have defined the roles of two autocrine/paracrine signaling mechanisms in ccRCC that control lipid homeostasis to promote renal cancer development. Because of the extracellular nature of the signal, interrupting lipid storage with toxic effects on the tumor cells becomes an attractive and seemingly feasible approach to develop a new line of anti-metabolic therapies for clear cell Renal Cell Cancer.
Supplementary Material
Statement of Significance.
Extracellular control of lipid accumulation via G-protein receptor-mediated cell signaling is a metabolic vulnerability in clear cell renal cell carcinoma, which depends on lipid storage to avoid oxidative toxicity.
Acknowledgements:
This work was supported by funding to D.B.L. (GM101171 and CA253986), D.G.I. (R35GM119518), and S.M.W. (CA254409, JEK Grant 21K07, and P30CA240139). The Metabolomics Core Facility at MD Anderson Cancer Center was supported in part by Cancer Prevention Research Institute of Texas [CPRIT] grant number RP130397 and NIH grants S10OD012304-01, U01CA235510, and P30CA016672. The Cancer Modeling Shared Resource (SCR022891), Flow Cytometry Shared Resource (SCR022501), and Oncogenomics Shared Resource (SCR022502) at the University of Miami Sylvester Comprehensive Cancer Center are supported P30CA240139.
References:
- 1.Farrukh M, Ali MA, Naveed M, Habib R, Khan H, Kashif T, et al. Efficacy and Safety of Checkpoint Inhibitors in Clear Cell Renal Cell Carcinoma: A Systematic Review of Clinical Trials. Hematology/oncology and stem cell therapy 2023;16:170–85 [DOI] [PubMed] [Google Scholar]
- 2.Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet 2009;373:1119–32 [DOI] [PubMed] [Google Scholar]
- 3.Barata PC, Rini BI. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J Clin 2017;67:507–24 [DOI] [PubMed] [Google Scholar]
- 4.Dell’Atti L, Bianchi N, Aguiari G. New Therapeutic Interventions for Kidney Carcinoma: Looking to the Future. Cancers 2022;14. [DOI] [PMC free article] [PubMed]
- 5.Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205 [DOI] [PubMed] [Google Scholar]
- 6.Safran M, Kaelin WG Jr. HIF hydroxylation and the mammalian oxygen-sensing pathway. J Clin Invest 2003;111:779–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 2012;33:207–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Mijn JC, Fu L, Khani F, Zhang T, Molina AM, Barbieri CE, et al. Combined Metabolomics and Genome-Wide Transcriptomics Analyses Show Multiple -Induced Changes in Lipid Metabolism in Early Stage Clear Cell Renal Cell Carcinoma. Transl Oncol 2020;13:177–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 2019;10:1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. -Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov 2015;5:652–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun 2017;8:1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz ALS, Barreto EA, Fazolini NPB, Viola JPB, Bozza PT. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis 2020;11:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan SK, Mahmud I, Fontanesi F, Puchowicz M, Neumann CKA, Griswold AJ, et al. Obesity-Dependent Adipokine Chemerin Suppresses Fatty Acid Oxidation to Confer Ferroptosis Resistance. Cancer Discov 2021;11:2072–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helfer G, Wu QF. Chemerin: a multifaceted adipokine involved in metabolic disorders. J Endocrinol 2018;238:R79–r94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rourke JL, Dranse HJ, Sinal CJ. Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes Rev 2013;14:245–62 [DOI] [PubMed] [Google Scholar]
- 16.Fang P, Han L, Yu M, Han S, Wang M, Huang Y, et al. Development of metabolic dysfunction in mice lacking chemerin. Molecular and Cellular Endocrinology 2021;535:111369. [DOI] [PubMed] [Google Scholar]
- 17.Yoshimura T, Oppenheim JJ. Chemokine-like receptor 1 (CMKLR1) and chemokine (C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp Cell Res 2011;317:674–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zabel BA, Allen SJ, Kulig P, Allen JA, Cichy J, Handel TM, et al. Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J Biol Chem 2005;280:34661–6 [DOI] [PubMed] [Google Scholar]
- 19.Ernst MC, Sinal CJ. Chemerin: at the crossroads of inflammation and obesity. Trends Endocrinol Metab 2010;21:660–7 [DOI] [PubMed] [Google Scholar]
- 20.Park JY, Kim MJ, Kim YK, Woo JS. Ceramide induces apoptosis via caspase-dependent and caspase-independent pathways in mesenchymal stem cells derived from human adipose tissue. Archives of toxicology 2011;85:1057–65 [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Murphy LO. Autophagy Assays for Biological Discovery and Therapeutic Development. Trends in biochemical sciences 2020;45:1080–93 [DOI] [PubMed] [Google Scholar]
- 22.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr 2007;27:79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvalho MV, Gonçalves-de-Albuquerque CF, Silva AR. PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. Int J Mol Sci 2021;22. [DOI] [PMC free article] [PubMed]
- 24.Noshiro M, Kawamoto T, Nakashima A, Ozaki N, Saeki M, Honda K, et al. DEC1 regulates the rhythmic expression of target genes involved in lipid metabolism in white adipose tissue. Genes to cells : devoted to molecular & cellular mechanisms 2020;25:232–41 [DOI] [PubMed] [Google Scholar]
- 25.Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell 2002;2:331–41 [DOI] [PubMed] [Google Scholar]
- 26.Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr 2014;34:281–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham KL, Zhang JV, Lewen S, Burke TM, Dang T, Zoudilova M, et al. A novel CMKLR1 small molecule antagonist suppresses CNS autoimmune inflammatory disease. Plos One 2014;9:e112925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elias R, Tcheuyap VT, Kaushik AK, Singla N, Gao M, Reig Torras O, et al. A renal cell carcinoma tumorgraft platform to advance precision medicine. Cell reports 2021;37:110055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med 2021;218. [DOI] [PMC free article] [PubMed]
- 30.Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016;16:732–49 [DOI] [PubMed] [Google Scholar]
- 31.Tun HW, Marlow LA, von Roemeling CA, Cooper SJ, Kreinest P, Wu K, et al. Pathway signature and cellular differentiation in clear cell renal cell carcinoma. Plos One 2010;5:e10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer letters 2018;435:92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao H, Mohamed EM, Xu GG, Waters M, Jing K, Ma Y, et al. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 2016;7:3832–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattern A, Zellmann T, Beck-Sickinger AG. Processing, signaling, and physiological function of chemerin. IUBMB life 2014;66:19–26 [DOI] [PubMed] [Google Scholar]
- 35.Fischer TF, Czerniak AS, Weiß T, Schoeder CT, Wolf P, Seitz O, et al. Ligand-binding and -scavenging of the chemerin receptor GPR1. Cellular and molecular life sciences : CMLS 2021;78:6265–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Henau O, Degroot GN, Imbault V, Robert V, De Poorter C, McHeik S, et al. Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. Plos One 2016;11:e0164179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samson M, Edinger AL, Stordeur P, Rucker J, Verhasselt V, Sharron M, et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. European journal of immunology 1998;28:1689–700 [DOI] [PubMed] [Google Scholar]
- 38.Bauer S, Wanninger J, Schmidhofer S, Weigert J, Neumeier M, Dorn C, et al. Sterol regulatory element-binding protein 2 (SREBP2) activation after excess triglyceride storage induces chemerin in hypertrophic adipocytes. Endocrinology 2011;152:26–35 [DOI] [PubMed] [Google Scholar]
- 39.Reichert CO, de Freitas FA, Levy D, Bydlowski SP. Chapter Fourteen - Oxysterols and mesenchymal stem cell biology. In: Litwack G, editor. Vitamins and Hormones: Academic Press; 2021. p 409–36. [DOI] [PubMed] [Google Scholar]
- 40.Pégorier JP, Le May C, Girard J. Control of gene expression by fatty acids. The Journal of nutrition 2004;134:2444s–9s [DOI] [PubMed] [Google Scholar]
- 41.Zheng C, Chen D, Zhang Y, Bai Y, Huang S, Zheng D, et al. FAM19A1 is a new ligand for GPR1 that modulates neural stem-cell proliferation and differentiation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2018:fj201800020RRR [DOI] [PubMed]
- 42.Schreiber R, Xie H, Schweiger M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochimica et biophysica acta Molecular and cell biology of lipids 2019;1864:880–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji ZS, Jiang H, Xie Y, Wei QP, Yin XF, Ye JH, et al. Chemerin promotes the pathogenesis of preeclampsia by activating CMKLR1/p-Akt/CEBPɑ axis and inducing M1 macrophage polarization. Cell biology and toxicology 2022;38:611–28 [DOI] [PubMed] [Google Scholar]
- 44.Tümmler C, Snapkov I, Wickström M, Moens U, Ljungblad L, Maria Elfman LH, et al. Inhibition of chemerin/CMKLR1 axis in neuroblastoma cells reduces clonogenicity and cell viability in vitro and impairs tumor growth in vivo. Oncotarget 2017;8:95135–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar V, LaJevic M, Pandrala M, Jacobo SA, Malhotra SV, Zabel BA. Novel CMKLR1 Inhibitors for Application in Demyelinating Disease. Sci Rep 2019;9:7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu J, Shen S, Liu T, Ren X, Zhu C, Liang Q, et al. Chemerin enhances mesenchymal features of glioblastoma by establishing autocrine and paracrine networks in a CMKLR1-dependent manner. Oncogene 2022;41:3024–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiao L, Wu X, Zhang J, Liu L, Sui X, Zhang R, et al. -NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2019;33:12760–7 [DOI] [PubMed] [Google Scholar]
- 48.Monjaras-Avila CU, Lorenzo-Leal AC, Luque-Badillo AC, D’Costa N, Chavez-Munoz C, Bach H. The Tumor Immune Microenvironment in Clear Cell Renal Cell Carcinoma. Int J Mol Sci 2023;24. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available upon request from the corresponding author. Lipidomic data are available at https://zenodo.org/records/10913324. RNAseq data generated in this study are publicly available at the EMBL ArrayExpress database with accession number E-MTAB-14016 (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-14016). Publicly available data analyzed in this study were obtained from the KIRC dataset of TCGA using GEPIA2 at http://gepia2.cancer-pku.cn/.