

Abstract
Regulation of the actin-activated ATPase of smooth muscle myosin II is known to involve an interaction between the two heads that is controlled by phosphorylation of the regulatory light chain. However, the three-dimensional structure of this inactivated form has been unknown. We have used a lipid monolayer to obtain two-dimensional crystalline arrays of the unphosphorylated inactive form of smooth muscle heavy meromyosin suitable for structural studies by electron cryomicroscopy of unstained, frozen-hydrated specimens. The three-dimensional structure reveals an asymmetric interaction between the two myosin heads. The ATPase activity of one head is sterically “blocked” because part of its actin-binding interface is positioned onto the converter domain of the second head. ATPase activity of the second head, which can bind actin, appears to be inhibited through stabilization of converter domain movements needed to release phosphate and achieve strong actin binding. When the subfragment 2 domain of heavy meromyosin is oriented as it would be in an actomyosin filament lattice, the position of the heads is very different from that needed to bind actin, suggesting an additional contribution to ATPase inhibition in situ.
Keywords: phosphorylation, 2-D crystalline arrays, myosin regulation, myosin light chains
Of the 15 types of myosin in the myosin superfamily, only isoforms of myosin II are capable of forming filaments (1). Myosin II consists of six polypeptide chains, two of which are heavy chains that contain actin-binding, ATP catalysis, and filament forming activities. Two pairs of light chains, an essential light chain (ELC) and a regulatory light chain (RLC), together with part of the heavy chain form a lever arm through which force is transmitted to produce filament sliding (2). Myosin II can be cleaved into several soluble subfragments. Myosin subfragment 1 (S1, the head portion of myosin) contains the ATPase and actin-binding regions of the heavy chain (also called the motor domain) and the light chain lever arm. Subfragment 2 (S2, the N-terminal portion of the myosin rod), which is predicted to have an α-helical coiled-coil structure (3), links S1 to the filament backbone and forms the myosin heavy chain dimerization interface. Another soluble subfragment, heavy meromyosin (HMM), consists of the two S1 heads and S2. Myosin IIs are found in all eukaryotic cells but are most prevalent in muscle cells, where they are assembled into an elaborate contractile apparatus.
The actin-activated ATPase of vertebrate striated muscle myosin II is regulated primarily by proteins bound to the actin filament. In contrast, the ATPase activity of smooth and nonmuscle myosin II is regulated by phosphorylation of S19 in the N-terminal region of the RLC (reviewed in ref. 4). The dephosphorylated form has low ATPase activity, which is greatly increased on phosphorylation (5). The structural basis of phosphorylation-dependent regulation in smooth muscle myosin has been studied extensively by using soluble subfragments. Two-headed fragments carrying the full S2 region retain phosphorylation-dependent regulation. If the S2 region is shortened, which weakens dimerization, regulation is lost (6). In addition, single-headed myosin and S1 alone are unregulated (7). These results implicate head–head interactions as a fundamental feature of regulation (8).
Although the biochemical effect of smooth muscle myosin II phosphorylation is well established, the structural basis of regulation is less well understood. Recently, we succeeded in forming two-dimensional arrays of both the phosphorylated and dephosphorylated forms of smooth muscle HMM on a positively charged lipid monolayer (9). The projection images of arrays of dephosphorylated HMM preserved in negative stain revealed an asymmetric interaction between the two S1 heads. This intramolecular head–head interaction was abolished when the RLC was thiophosphorylated. This paper reports on the three-dimensional (3D) structure of dephosphorylated smooth muscle HMM determined by electron cryomicroscopy of unstained frozen hydrated specimens. The present work facilitated the construction of a considerably better molecular model than was possible from the projection in negative stain, which lacks information on the relative placement of features perpendicular to the projection direction. The new molecular model reveals the proximity of interacting surface loops. The structure also reveals, to our knowledge for the first time in a 3D image, a segment of the S2 domain in proximity to the two heads, the location of which is correlated with earlier electron microscopic images. The proposed structure of the inhibited form of smooth muscle myosin is supported by a large volume of mutational experiments, which can now be correlated with a 3D structure.
Materials and Methods
Chicken gizzard smooth muscle HMM was expressed and isolated as described previously (9). Crystallization was carried out at 4°C on a positively charged lipid monolayer system (10). Crystallization buffers consisted of 1 mM Mg2+, 20 mM phosphate, pH range 7–8, 1 mM ATP, 1 mM EGTA and polyethylene glycol 6000 (7–10%), and NaCl (90–120 mM). Specimens were plunge-frozen for cryoelectron microscopy as described (11) and examined on a Philips (Eindhoven, The Netherlands) CM300-FEG electron microscope at 300 kV accelerating voltage and at ×24,000 by using low-dose procedures. Micrographs were screened initially by optical diffraction and digitized on a Perkin–Elmer PDS1010 M microdensitometer at a step size of 0.54 nm with respect to the original object.
Micrographs were processed by using the ICE image processing software (12). Crystal unbending (13) and merging was done as previously described (9). The 3D reconstruction was obtained by merging the structure factors of 101 images tilted to as high as 60° by standard methods (14). The average unit cell dimensions derived from images of untilted specimens are a = 13.3 ± 0.2 nm, b = 30.4 ± 0.9 nm, c = 9.0 nm, and γ = 91.5° ± 0.8°, giving a resolution of 2.0 nm. Structure factors were refined to a common phase origin and merged in the two-sided plane group p2. The averaged phase residual was 25°.
Model Building.
For model building, we constructed an HMM atomic model starting with the x-ray crystal structure of the smooth muscle motor domain plus ELC (15) (PDB ID code 1BR1). Because this structure lacks the RLC, we used the corresponding segments of the x-ray structure of chicken skeletal myosin S1 (PDB ID code 2MYS) as a homology model of this feature (16). For alignment, we used the homologous sequences of the ELC to position the skeletal RLC and missing portions of the heavy chain into the model. We did not construct a homology model for the smooth muscle RLC, which has 55.6% sequence identity with the skeletal RLC. The HMM construct also contains heavy chain residues 853-1175, which comprise the S2 subfragment. This S2 portion was modeled by using an α-helical coiled-coil simulation program kindly provided by G. Offer (University of Bristol, Bristol, U.K.) (17). In total, 91 residues for each myosin heavy chain were built into the model to represent the S2 segment. Models were built to fit the electron density by using the x-ray crystallography modeling program o (18).
Results
Electron micrographs of the unstained frozen-hydrated two-dimensional crystals of unphosphorylated HMM are virtually featureless, but their Fourier transforms reveal strong diffraction spots to a resolution of ≈2.0 nm, which is comparable to the resolution obtained in negative stain (9). Image processing showed strong P2 symmetry, indicative of the presence of two HMM molecules within the unit cell (Fig. 1A). A 2-fold axis perpendicular to the plane of the crystal relates the pair of HMM molecules in the unit cell, but there is no symmetry relationship between the two heads within a single molecule.
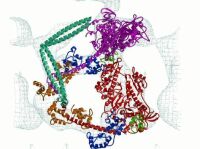
Figure 1.
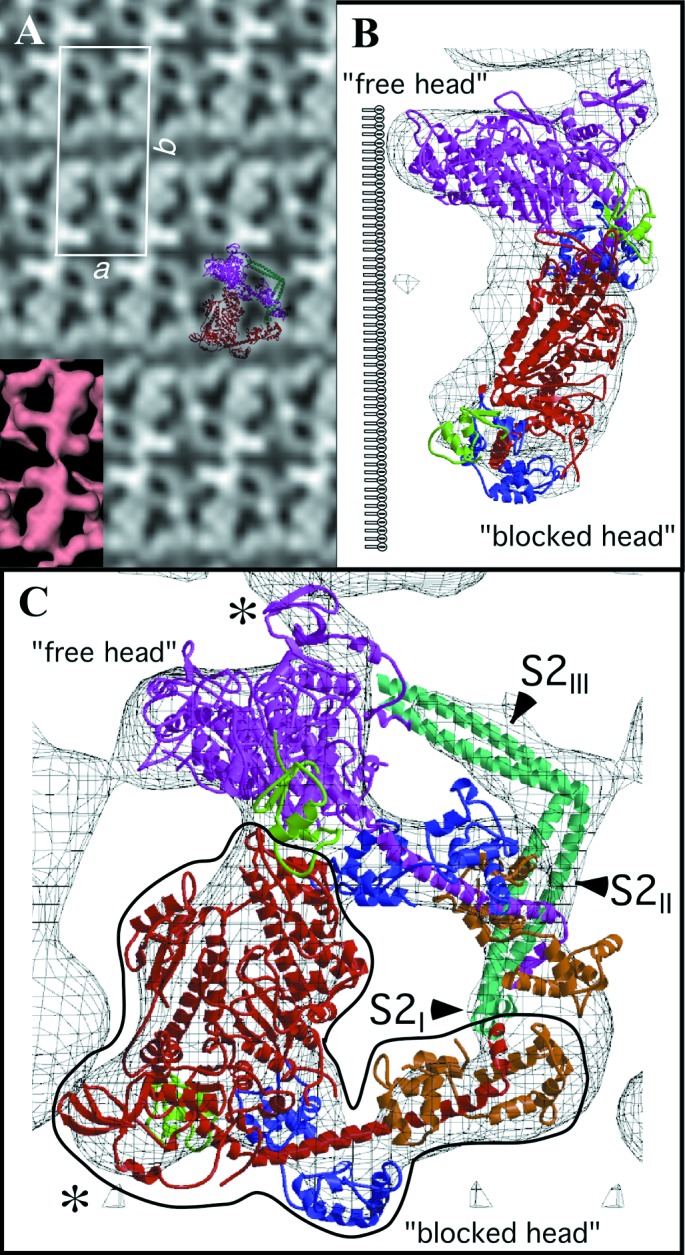
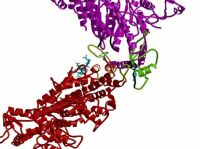
Two-dimensional arrays of unphosphorylated smooth muscle HMM. (A) Averaged in-plane projection of frozen hydrated arrays with a surface view of the 3D reconstruction as Inset (Lower Left). Overlaid on the projection is a view of the 3D model reduced to the same scale. The two S1s are colored red and magenta and the S2 cyan. (B and C) Two views of the model fitted to the reconstruction. B is oriented along the crystal a axis. The location of the lipid monolayer, which is not seen in the reconstruction, is drawn to the left of the structure. C is oriented perpendicular to the plane of the crystal. The lipid monolayer would be located below the structure in this view. The “blocked” head is outlined in black. The color scheme for all ribbon diagrams is as follows: the heavy chain of the “free” myosin head is magenta, and that of the “blocked” head is red. For both myosin heads, the converter domain is green, the ELCs blue, and the RLCs orange. Figures were prepared by using bobscript and raster3d (41, 42). The region displayed in C can be seen in stereo in Fig. 5 as well as Movie 1, which are published as supplemental data on the PNAS web site, www.pnas.org.
The 3D reconstruction reveals a complicated shape, but the map can be interpreted by model building. Because no atomic structure exists for a two-headed myosin II species, we built an HMM model as described above and fit it manually to the reconstruction envelope. The motor domains can be docked unambiguously into the electron density because of the asymmetry provided by the SH3 domain (Fig. 1B). The light chain lever arm of one S1 head, dubbed the “blocked” head, fits within the density without alterations, but that of the second head, the “free” head, required repositioning. We used heavy chain residue I796 between the ELC and converter as the pivot point for a rigid body rotation of ≈20° to position the model within the density. This movement, when referred to the direction of force production on an actin filament, is almost entirely azimuthal in direction and suggests lever arm flexibility in this direction as a characteristic of myosin structure.
The most surprising feature of the arrangement of heads within HMM is the asymmetry (Fig. 1 B and C). Part of the actin-binding interface of the “blocked” head (the upper 50-kDa domain) bridges the “free” head catalytic and converter domains as well as its ELC. The converter domain (residues 719–780) is believed to function as a structure that transmits conformational changes in the ATP-binding site to the lever arm (19). Two surfaces of the motor domain are involved in stabilizing this interaction (Fig. 2 A and B). Loop 407–417 on the “blocked” head is located near residue 458 and loop 167–170 of the “free head” catalytic domain. Residue 458 is located at the start of a β-strand that connects to switch 2 [residues 468–508 (20)]. In addition, “blocked” head loop 368–379 is positioned over helix 727–732 and loop 746–749 of the “free” head converter domain. Finally, loop 615–618 and helix 392–398 of the “blocked” head are in close proximity with residues 98–101, which connect the E and F helices of the “free” head ELC. These intramolecular head interactions are abolished when HMM is activated by thiophosphorylation (9).
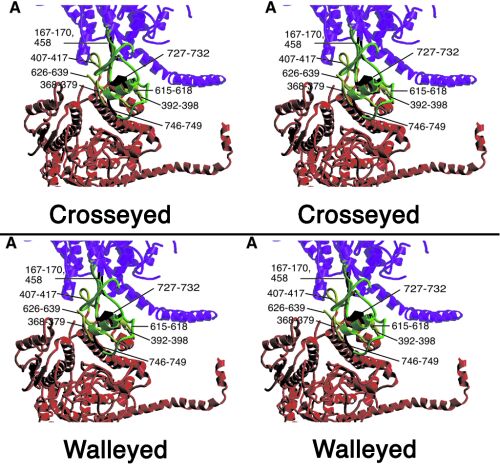
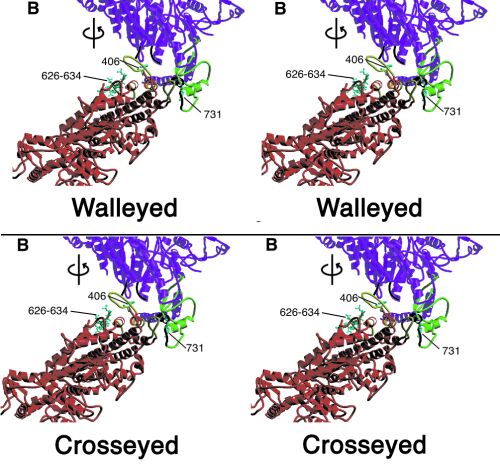
Figure 2.
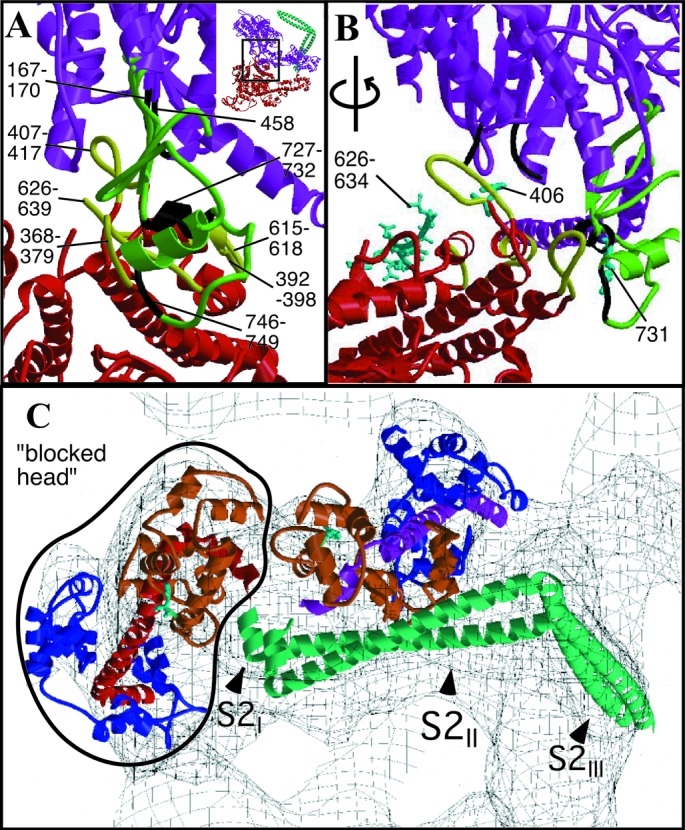
(A and B) Heavy chain–heavy chain interactions. Color scheme as in Fig. 1, except that loops involved in heavy chain–heavy chain interactions are in black on the “free” head and in yellow on the “blocked” head. (A) View perpendicular to the crystal plane (upper right-hand corner Inset shows corresponding overall view). “Free” head residues potentially involved in the interaction include 458 (at the end of a disordered loop), loop 167–170, and converter domain loop 746–749 and helix 727–732. “Blocked” head residues include loop 368–379, the myopathy loop 407–417, loop 615–618, and helix 392–398. (B) Same region rotated by ≈70° to view within the crystal plane. Displayed in cyan in ball-and-stick format are residues 626–634 of the smooth muscle inhibitory domain (43) and “blocked” head residue 406 and “free” head residue 731. (C) S1–S2 junction viewed down the a axis of the unit cell. The light chain lever arm of the “blocked” head is outlined in black. The N termini of the RLCs (F19, smooth muscle F25) are shown in cyan as ball-and-stick structures. Figs. 6–8 are stereoviews of Fig. 2 A–C; the region shown in Fig. 2 A and B is Movie 2; Fig 2C is Movie 3. Figs. 6–8 and Movies 2 and 3 are published as supplemental data on the PNAS web site, www.pnas.org.
Close to the S1-S2 junction, the resolution is insufficient to resolve the initial part of the S2 domain from the light chain lever arm, so that the relative position of the C-terminal 839–852 helix of S1 is somewhat ambiguous. We built two models that differed in this region by the way the light chain lever arm of the “free” head was modified (the “blocked” head was never modified). In the model illustrated here, no attempt was made to symmetrize the S1–S2 junction. Consequently, the alterations in the light chain lever arm are simpler, requiring only a single pivot point and a single rotation. This model also maintains the orientation of the 839–852 helix of the “free” head relative to the direction of force production in a thick filament the same as in the Rayment et al. acto-S1 structure (2). In the other model (not illustrated), we attempted to symmetrize the S1–S2 junction around a local 2-fold axis, which required more severe modification to the light chain lever arm. From both models, however, the structural features of the S2 segment that we infer are similar.
In addition to the motor domain and light chain lever arms, the reconstruction shows density that cannot be attributed to the S1 and therefore must correspond to the S2 domain. Our expressed HMM contains sufficient sequence to form 48 nm of S2. The reconstruction shows about 8.8 nm of density that can be attributed to S2 and a further 4.7 nm that can be inferred (Fig. 1B). The remaining 34-nm C-terminal portion of S2 is not resolved and is probably disordered. On the basis of secondary structure predictions (3), we have built S2 into the model as completely α-helical and mostly coiled-coil in three segments separated by sharp bends. The C-terminal segment, S2III, shows the left-handed twist predicted for an α-helical coiled coil and can be modeled with 40 residues of coiled coil. The next segment, S2II, is only partly resolved but extends at an angle of almost 90° off of S2III. The visible part of S2II can be modeled with 19 residues of coiled coil. These two visible parts of S2 are spatially distant from the S1-S2 junction and would require about 4.7 nm of α-helix (≈32 residues) to form a connection.
The connecting segment of S2 was modeled in two parts. Since this connector is not resolved because of its close proximity to the “free” head RLC, its structure is somewhat based on conjecture. Nevertheless, some structural element must connect the visible portion of S2 with the C terminus of S1. The model in this case was based on two observations. First, the S2ll segment cannot be extended directly to the C terminus of S1; some kind of bend is needed to make the connection. Second, the connection was formed by simply incorporating a 90° turn similar to that found at the junction between S2ll and S2lll. The total length of S2ll is 6.2 nm (≈43 residues), including the visible part and the extension. The segment (labeled S2l in Fig. 1C, cyan), which connects S2ll to S1, consists of 8 amino acids of α-helix that is distorted from the standard coiled-coil configuration. The N-terminal Cαs of S2l are within 0.36 and 0.47 nm of the K852 Cα of S1. Note that the S2l segment is not a simple extension of the 839–852 heavy chain helix but extends from it at a sharp angle, thus implying three breaks in the α-helical structure at the beginning of S2.
The direction of view shown in Fig. 1 A and C places the monolayer below the HMM and in Fig. 1B to the left of HMM. Contact with the monolayer occurs primarily through S2 and the actin-binding surface of the “free” head. Most of the HMM mass, including the “blocked” head, appears to be held away from the monolayer. The HMM–monolayer interaction is primarily because of opposite net charge; the monolayer is positively charged independent of pH (10). Binding of HMM is most likely mediated by the S2 domain (pI 4.7), because the surface residues of the “free” head in contact with the monolayer (residues 368–386, 392–418, and 298–309) collectively have a pI of 10.2 and are unlikely to be electrostatically attracted to the monolayer. Because most of the HMM is distant from the monolayer, it seems unlikely that the monolayer is enforcing a particular conformation on the molecule. The orientation of the “free” head in particular is more likely to be determined by interactions within the HMM than by interactions with the monolayer.
Discussion
The present work provides, to our knowledge, the first 3D image of the inhibited state of smooth muscle myosin. Although the image is of modest resolution by x-ray crystallography standards, the availability of x-ray structures of both the smooth muscle motor domain plus ELC and the skeletal muscle S1 with intact lever arm makes it possible to derive an atomic model of the inhibited state of smooth muscle HMM that is supported by a considerable body of evidence. Previous experience has shown that atomic models can be docked into 2-nm-resolution electron microscopy reconstructions with a precision of ≈0.5 nm, which is good enough to identify interacting loops (21).
The present HMM structure is considerably different from the only previous model for an HMM, that of scallop striated muscle, which contains two S1 heads symmetrically placed around a 2-fold rotation axis at the beginning of S2 (17). We attempted to build a model with symmetry at the S1-S2 junction, but this required a torsional rotation by almost 90° of the “free” head RLC and its associated heavy chain about heavy chain residue G821 to bring the C-terminal α-helices of the S1s (residues 839–852) into a 2-fold relationship. However, there is no compelling reason to enforce this symmetry, because the initial direction that S2l most probably takes from this point is nearly perpendicular to any 2-fold axis that can be built into the model (Fig. 2C). That is to say, the symmetry built into the model cannot be propagated beyond the C termini of S1. We therefore favor a model in which there is no region of symmetry between the two S1s.
We also attempted to construct our HMM model by using the x-ray structure of the scallop light chain lever arm (22) in place of the chicken skeletal structure. Although scallop myosin is regulated by its RLC (23), the crystal structure is of the Ca2+-bound “on” state. The scallop structure was a poor fit because it is less bent in the heavy chain between the ELC and the RLC. The chicken skeletal structure, in contrast, required no modification other than the one rotation within the “free” head described earlier. This observation supports previous suggestions that one aspect of inactivation involves changes in flexibility of the light chain lever arm that would facilitate head–head interactions (22).
Our HMM construct contains a complete S2 domain, only part of which is directly visible in the reconstruction. However, of the 91 residues of S2 built into the model, 59 are located in clearly resolved features, and the remaining 32, which are located in density associated with the “free” head RLC, were added to connect the visible part of S2 with the C termini of S1. Only the S2III segment shows evidence of a coiled-coil structure, because it shows a left-handed twist. The S2II segment, which was built as a coiled-coil on the basis of structure prediction rather than observation, may not form a coiled coil, although it is most probably α-helical. The modeled segment of S2, which passes very close to the “free” head RLC, suggests specific interactions with the S1 heads that stabilize this segment, thereby making S2 visible in the averaged reconstruction.
It seems certain that in the S2 part of the reconstruction, there are clear departures from a strict coiled-coil structure in the form of sharp bends. One 90° bend is directly visible at the junction between S2II and S2III. Two other bends are inferred, one at the S1–S2 junction and another at the junction between S2I and S2II. These bends at the beginning of S2 imply weak points in the coiled-coil interaction in this region. Studies have shown that expressed HMM constructs containing less than 25 heptads of native rod sequence show reduced dimerization and therefore reduced regulation (24). The 91 residues of S2 built into the model (13 heptads) are well short of the minimal number needed to form stable dimers and therefore confer complete regulation on the HMM (6).
Although dimerization is essential for complete regulation, other interactions involving the S2 domain are also essential. When a leucine zipper (32 amino acids) is incorporated after 0, 2, or 7 heptads of native S2 sequence, regulation is only partially obtained. Only when the leucine zipper is incorporated after 15 heptads of native rod sequence is regulation complete. These studies suggest that full regulation requires not only dimerization but also an interaction between the myosin heads and the initial S2 sequence. In addition, HMM engineered to contain a leucine zipper near the S1–S2 junction is only partially regulated and produces smaller unitary displacements than wild-type HMM, implying that some structural weakness in the coiled coil is necessary for optimal myosin function.§ The HMM model shown here supports both of these conclusions by indicating close proximity between S2 and the “free” S1 head as well as breaks in the coiled-coil structure that imply weakness in the initial S2 segment.
The structure obtained here is unusual because of the asymmetry of the interaction between two otherwise identical molecules. Before this study, the only evidence for the structure of dephosphorylated smooth muscle HMM came from rotary shadowing electron microscopy of isolated molecules (25). Typically, these preparations reveal a conformation with the heads folded backwards toward the S2 domain (26). Our structure supports this conformation, because the S2IlI segment extends backwards in the same direction that the S1 heads are folded back. However, previously published structures of double-headed myosins imaged by rotary shadowing mostly show the heads separated, whereas our model shows them interacting to form a compact structure. The conditions normally used for rotary shadowing apply considerable stress to weak protein–protein interactions. It is known, for example, that regulation is highly ionic strength-dependent (8), and it is increasing ionic strength to which HMM is subjected during rotary shadowing specimen preparation. These effects can be somewhat offset by using peptide crosslinkers, and in those few instances where a crosslinker has been used, a clearly more compact arrangement of the myosin heads can be detected, as predicted by our model (25, 27).
Several studies using mutated smooth muscle HMM as well as chimeric constructs that were previously rather puzzling are explained by our structure. It is well known that regulation requires a two-headed species of myosin II (7, 8), but it was unknown whether complete heads were needed or whether a complete head in combination with a light chain domain would be sufficient. This question has been addressed in two recent studies using HMM constructs with one complete S1 head and a second complete light chain domain. Sweeney et al. (28) concluded that a high degree of regulation can be obtained only with two intact heads, a feature that is a requirement of the structural model proposed here. The study done by Ikebe and colleagues (29) reached the opposite conclusion, namely that two RLC domains are sufficient for regulation. The latter study, however, chose assay conditions that precluded an accurate discrimination between complete versus partial regulation.
The converter domain is a key element of the interacting surfaces of the two S1 heads. Substitution of the converter domain of smooth muscle HMM with that from skeletal muscle produces an unregulated HMM (30). However, substitution of the skeletal ELC for the smooth ELC has no effect on regulation (24). In our structure, the “blocked” head interacts with the ELC loop 97–101 of the “free” head. This sequence is identical in the ELC of both smooth and skeletal muscle.
Yamashita et al. (31) characterized point mutations in the smooth muscle myosin heavy chain that correspond to loci known to cause familial hypertrophic cardiomyopathy. One of these mutations, R406Q (cardiac R403Q), caused partial loss of regulation. This residue is in one of the loops of the “blocked” head that lies near “free” head residues 458 and 167–170 in our structure and extends in the direction of the “free” head. More significantly, a mutation in the converter domain, R731W (cardiac R719W) causes nearly complete loss of regulation but is wild type in other characteristics. R731 in the “free” head is in a helix positioned close to the 368–379 loop of the “blocked” head.
Chimeric substitutions of the actin-binding loop (HC residues 626–653) with the corresponding loop from skeletal muscle myosin produced a molecule enzymatically active in the dephosphorylated state (32). In our structure, the part of this insert that is visible in the “blocked” head (residues 626–634) is located immediately behind the surface that interacts with the “free” head. The rest of the actin-binding loop, although not visible, would likely be in proximity to the “free” head. These studies indicate that interaction between the two myosin heads is required to obtain the inhibited state, and that the key regions that are interacting are the converter domain and part of the actin-binding interface.
In smooth muscle, phosphorylation of the RLC is required to activate the actin-activated ATPase. In the inhibited state, the RLCs of adjacent heads can be crosslinked to each other, and this crosslinking is abolished in the activated state (33). However, other work has shown that deletion of the first 16 N-terminal residues of the RLC produces a myosin whose actin-activated ATPase cannot be activated by phosphorylation (34). Although crosslinking results indicate that the inhibited state involves an RLC–RLC interaction, phosphorylation not only abolishes it, but must also promote binding of the N-terminal peptide with the rest of the RLC or with other parts of the light chain lever arm.
The RLC region of the x-ray structure of skeletal muscle S1, on which our atomic model is based, extends only to F19 (F25 in smooth muscle). Twenty-four amino acids are thus unaccounted for in our model structure. The reconstruction provides no direct evidence of the location of this peptide, which would not stand out at our resolution unless it had some secondary structure and was well separated from nearby structures. Because of the asymmetric arrangement of the S1s, the N termini of the two RLCs are in very different locations (Fig. 2C, cyan residues). On the “blocked” head, F19 is located to the outside of the structure away from the “free” head, a location that would be unfavorable for interaction with the “free” head. For the “free” head RLC, F19 is located on the side proximal to the “blocked” head RLC, a location that would be favorable for RLC–RLC crosslinking. The asymmetric location of the N-terminal peptides suggests the possibility that they can be involved in more than one kind of interaction in the dephosphorylated state. One such possibility is involvement in formation of the 10S structure in smooth muscle myosin, which cannot be formed in the absence of the RLC N-terminal peptide (34).
There are two aspects to the regulation of ATPase activity in smooth muscle myosin. Dephosphorylation reduces the ATPase activity by ≈200-fold, but binding to actin is reduced only ≈4-fold, leading to the proposal that phosphate release is the step in the ATPase cycle that is regulated (5). The small decrease in binding affinity on dephosphorylation may reflect a change from double-headed to single-headed binding. For this to be true, one head of HMM must retain actin-binding capability. Our reconstruction offers an explanation of both features of smooth muscle regulation.
In our model, the “blocked” head cannot bind actin, because part of its actin-binding interface is interacting with the converter domain of the “free” head. When the “blocked” head is aligned on actin with the rigor actomyosin model (35) on the basis of sequence similarities in the motor domain (Fig. 3A Inset), about half of the “free” head catalytic domain overlaps the actin filament. When the “free” head of HMM is similarly aligned to rigor actoS1, the “free” head fits without the “blocked” head clashing with the actin filament (Fig. 3A). Considerable space separates the HMM from the actin filament to accommodate the possibility that the orientation of the myosin head in the weak binding state differs significantly from rigor, as suggested by 3D images obtained of contracting muscle (36). The orientation of the light chain lever arm in the actin-bound “free” head resembles that predicted for the conformation at the beginning of the powerstroke (reviewed in refs. 15, 37, and 38), supporting the idea that these heads are in a state that precedes phosphate release. Thus, the form of HMM seen in our crystals could, in principle, be capable of binding actin with one head.
Figure 3.
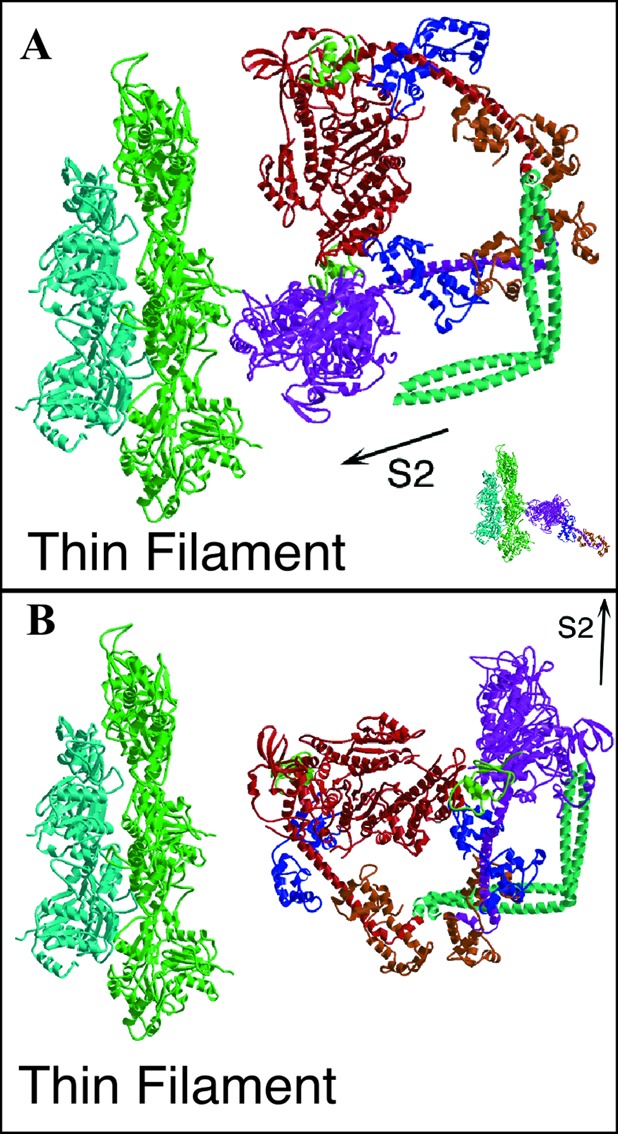
Docking of unphosphorylated HMM onto actin. (A) The “free” head of the HMM model is oriented on actin by using the actin-binding domain of rigor skeletal muscle actomyosin (Inset) as the reference. The smooth muscle S1 with a transition state analog at the active site appears with the lever arm up (15) in contrast to the lever arm down-postpowerstroke conformation of skeletal muscle actomyosin (35). The HMM model, when oriented via the actin-binding domain, verifies that one head can interact with actin without steric hindrance from the second head. The orientation of the double-headed myosin in B represents the arrangement with the S2lll segment oriented toward the direction of force transmission, as would occur within the muscle lattice. The head orientations in this configuration do not favor actin binding by either head.
Because the “free” head can bind actin, its ATPase inhibition must be achieved by a different mechanism. It is widely believed that structural changes involving the converter domain are a necessary prerequisite for the release of phosphate through myosin's “back door” (39). We believe that the binding of a single domain of the “blocked” head, i.e., the upper 50 kDa across three domains of the “free” head that are known to move relative to one another stabilizes the “free” head converter domain against the movements necessary to open the back door, accelerate phosphate release, and enter the strong binding conformation. Thus, ATPase inactivation can be achieved for both heads by using different mechanisms.
The force produced by the myosin head is ultimately transmitted to the thick filament via the S2 segment. Somewhat surprisingly, the alignment of HMM on actin positions the S2lll segment away from the direction that force would be transmitted during an active contraction (Fig. 3A). This could mean two things. One possibility is simply that the S1–S2 junction is flexible, and that the structure formed in the crystal is the one most compatible with the head packing. However, the orientation of the heads in rotary shadowed HMM argues against this possibility. Alternatively, this orientation of the S2 may be specific for the inhibited state of smooth muscle myosin in filaments. The effect can be illustrated by orienting the S2lll segment in the direction it would have if present in a thick filament (Fig. 3B). In this orientation, the myosin heads in dephosphorylated thick filaments would be rotated ≈90° away from the orientation needed to bind actin. In an oriented filament array, especially the side polar myosin filaments of smooth muscle (40), this would constitute a significant barrier for actomyosin interactions (Fig. 3B). Thus, in the filamentous state, this sterically unfavorable head arrangement may be an important component of regulation, unlike the modest change in phosphorylation-dependent binding seen with soluble HMM.
These observations can be combined into a scheme for the “on” switch in smooth muscle myosin II (Fig. 4). Unphosphorylated myosin forms the head–head interaction where even the “free” head is bent backwards toward the thick filament backbone and oriented away from the thin filament, thereby decreasing both actin-binding and ATPase rate (Fig. 4A). On RLC phosphorylation, the “blocked” and “free” head separate and can independently search for actin monomers in a favorable orientation for myosin binding (Fig. 4B). When actin binding occurs (Fig. 4C), the initial state is configured with the light chain lever arm oriented toward the pointed end of actin in the prepowerstroke position (upper configuration), which, with filament sliding, will rotate toward the barbed end at the end of the powerstroke (lower configuration).
Figure 4.
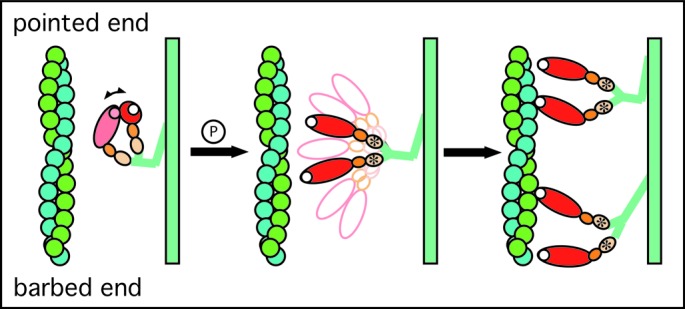
Diagram illustrating a model for the structural changes that occur on activation of smooth muscle myosin. (A) In the “off” state, the two heads of myosin are in an orientation that is disadvantageous for actin binding by either head. (B) On phosphorylation of the RLC, the head–head interaction is interrupted, and both can search independently for actin monomers suitably placed for binding. (C) Myosin heads bind actin in the prepowerstroke position, which on filament sliding will transform to the rigor-like configuration that characterizes the end of the powerstroke. Note that the initial attachment to actin is likely to be considerably less ordered than implied by the cartoon (36).
The current report gives, to our knowledge, the first 3D view of the head–head interactions that are key to the regulation of smooth muscle myosin ATPase activity. Further work may establish the generality of this structure to myosin linked ATPase regulation in other systems.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants to K. Trybus and K. Taylor. We acknowledge the assistance of Mr. Gary Connors in the production of the supplemental movie material. We are also grateful to Dr. Lee Sweeney for helpful discussions and to Dr. Gerald Offer for providing us with a copy of his coiled-coil structure-generating program.
Abbreviations
- HMM
heavy meromyosin
- S1
subfragment 1, the head portion of myosin
- S2
subfragment 2, the N-terminal portion of the myosin rod
- RLC
regulatory light chain
- ELC
essential light chain
- 3D
three-dimensional
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1I84).
Lauzon, A., Warshaw, D. M. & Trybus, K. M. (2000) Biophys. J. 78, 243A (abstr.).
References
- 1.Sellers J R. In: Protein Profile. Sheterline P, editor. Oxford, U.K.: Oxford Univ. Press; 1999. [Google Scholar]
- 2.Rayment I, Holden H M, Whittaker M, Yohn C B, Lorenz M, Holmes K C, Milligan R A. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 3.McLachlan A D, Karn J. J Mol Biol. 1983;164:605–626. doi: 10.1016/0022-2836(83)90053-0. [DOI] [PubMed] [Google Scholar]
- 4.Sellers J R. Curr Opin Cell Biol. 1991;3:98–104. doi: 10.1016/0955-0674(91)90171-t. [DOI] [PubMed] [Google Scholar]
- 5.Sellers J R. J Biol Chem. 1985;260:15815–15819. [PubMed] [Google Scholar]
- 6.Trybus K M, Freyzon Y, Faust L Z, Sweeney H L. Proc Natl Acad Sci USA. 1997;94:48–52. doi: 10.1073/pnas.94.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cremo C R, Sellers J R, Facemyer K C. J Biol Chem. 1995;270:2171–2175. doi: 10.1074/jbc.270.5.2171. [DOI] [PubMed] [Google Scholar]
- 8.Ikebe M, Hartshorne D J. Biochemistry. 1985;24:2380–2387. doi: 10.1021/bi00330a038. [DOI] [PubMed] [Google Scholar]
- 9.Wendt T, Taylor D, Messier T, Trybus K, Taylor K. J Cell Biol. 1999;147:1–5. doi: 10.1083/jcb.147.7.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor K A, Taylor D W. J Struct Biol. 1992;108:140–147. doi: 10.1016/1047-8477(92)90013-z. [DOI] [PubMed] [Google Scholar]
- 11.Dubochet J, Adrian M, Chang J J, Homo J C, Lepault J, McDowall A W, Schultz P. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 12.Hardt S, Wang B, Schmid M. J Struct Biol. 1996;116:68–70. doi: 10.1006/jsbi.1996.0012. [DOI] [PubMed] [Google Scholar]
- 13.Henderson R, Baldwin J M, Downing K H, Lepault J, Zemlin F. Ultramicroscopy. 1986;19:147–178. [Google Scholar]
- 14.Amos L A, Henderson R, Unwin P N T. Prog Biophys Mol Biol. 1982;39:183–231. doi: 10.1016/0079-6107(83)90017-2. [DOI] [PubMed] [Google Scholar]
- 15.Dominguez R, Freyzon Y, Trybus K M, Cohen C. Cell. 1998;94:559–571. doi: 10.1016/s0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- 16.Rayment I, Rypniewski W R, Schmid-Bäse K, Smith R, Tomchick D R, Benning M M, Winkelmann D A, Wesenberg G, Holden H M. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 17.Offer G, Knight P. J Mol Biol. 1996;256:407–416. doi: 10.1006/jmbi.1996.0096. [DOI] [PubMed] [Google Scholar]
- 18.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 19.Houdusse A, Kalabokis V, Himmel D, Szent-Gyorgyi A, Cohen C. Cell. 1999;97:459–470. doi: 10.1016/s0092-8674(00)80756-4. [DOI] [PubMed] [Google Scholar]
- 20.Cooke R. Physiol Rev. 1997;77:671–697. doi: 10.1152/physrev.1997.77.3.671. [DOI] [PubMed] [Google Scholar]
- 21.Baker T S, Johnson J E. Curr Opin Struct Biol. 1996;6:585–594. doi: 10.1016/s0959-440x(96)80023-6. [DOI] [PubMed] [Google Scholar]
- 22.Houdusse A, Cohen C. Structure (London) 1996;4:21–32. doi: 10.1016/s0969-2126(96)00006-8. [DOI] [PubMed] [Google Scholar]
- 23.Szent-Gyorgyi A G, Kalabokis V N, Perreault-Micale C L. Mol Cell Biochem. 1999;190:55–62. [PubMed] [Google Scholar]
- 24.Trybus K M. J Biol Chem. 1994;269:20819–20822. [PubMed] [Google Scholar]
- 25.Trybus K M, Huiatt T W, Lowey S. Proc Natl Acad Sci USA. 1982;79:6151–6155. doi: 10.1073/pnas.79.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H, Stafford W F, Slayter H S, Seidel J C. J Biol Chem. 1985;260:14810–14817. [PubMed] [Google Scholar]
- 27.Trybus K M, Lowey S. J Biol Chem. 1985;260:15988–15995. [PubMed] [Google Scholar]
- 28.Sweeney H L, Chen L Q, Trybus K M. J Biol Chem. 2000;275:41273–41277. doi: 10.1074/jbc.M008310200. [DOI] [PubMed] [Google Scholar]
- 29.Li X D, Saito J, Ikebe R, Mabuchi K, Ikebe M. Biochemistry. 2000;39:2254–2260. doi: 10.1021/bi9924617. [DOI] [PubMed] [Google Scholar]
- 30.Trybus K M, Naroditskaya V, Sweeney H L. J Biol Chem. 1998;273:18423–18428. doi: 10.1074/jbc.273.29.18423. [DOI] [PubMed] [Google Scholar]
- 31.Yamashita H, Tyska M J, Warshaw D M, Lowey S, Trybus K M. J Biol Chem. 2000;275:28045–28052. doi: 10.1074/jbc.M005485200. [DOI] [PubMed] [Google Scholar]
- 32.Rovner A S, Freyzon Y, Trybus K M. J Biol Chem. 1995;270:30260–30263. doi: 10.1074/jbc.270.51.30260. [DOI] [PubMed] [Google Scholar]
- 33.Wu X, Clack B A, Zhi G, Stull J T, Cremo C R. J Biol Chem. 1999;274:20328–20335. doi: 10.1074/jbc.274.29.20328. [DOI] [PubMed] [Google Scholar]
- 34.Ikebe M, Ikebe R, Kamisoyama H, Reardon S, Schwonek J P, Sanders C R, 2nd, Matsuura M. J Biol Chem. 1994;269:28173–28180. [PubMed] [Google Scholar]
- 35.Mendelson R, Morris E P. Proc Natl Acad Sci USA. 1997;94:8533–8538. doi: 10.1073/pnas.94.16.8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor K A, Schmitz H, Reedy M C, Goldman Y E, Franzini-Armstrong C, Sasaki H, Tregear R T, Poole K, Lucaveche C, Edwards R J, et al. Cell. 1999;99:421–431. doi: 10.1016/s0092-8674(00)81528-7. [DOI] [PubMed] [Google Scholar]
- 37.Cooke R. Proc Natl Acad Sci USA. 1998;95:2720–2722. doi: 10.1073/pnas.95.6.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmes K C. Curr Biol. 1997;7:R112–R118. doi: 10.1016/s0960-9822(06)00051-0. [DOI] [PubMed] [Google Scholar]
- 39.Yount R G, Lawson D, Rayment I. Biophys J. 1995;68:44s–49s. [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J-Q, Harder B A, Uman P, Craig R. J Cell Biol. 1996;134:53–66. doi: 10.1083/jcb.134.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merritt E A, Bacon D J. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 42.Esnouf R M. J Mol Graphics Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 43.Rovner A S. J Biol Chem. 1998;273:27939–27944. doi: 10.1074/jbc.273.43.27939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}