

Abstract
Down syndrome (trisomy 21) is the most common viable chromosomal disorder with intellectual impairment and several other developmental abnormalities. Here, we report the generation and characterization of induced pluripotent stem cells (iPSCs) derived from monozygotic twins discordant for trisomy 21 in order to eliminate the effects of the variability of genomic background. The alterations observed by genetic analysis at the iPSC level and at first approximation in early development illustrate the developmental disease transcriptional signature of Down syndrome. Moreover, we observed an abnormal neural differentiation of Down syndrome iPSCs in vivo when formed teratoma in NOD-SCID mice, and in vitro when differentiated into neuroprogenitors and neurons. These defects were associated with changes in the architecture and density of neurons, astroglial and oligodendroglial cells together with misexpression of genes involved in neurogenesis, lineage specification and differentiation. Furthermore, we provide novel evidence that dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A ( DYRK1A) on chromosome 21 likely contributes to these defects. Importantly, we found that targeting DYRK1A pharmacologically or by shRNA results in a considerable correction of these defects.
Keywords: disease modelling, Down syndrome, DYRK1A, induced pluripotent stem cells, neurodevelopment
Introduction
Down syndrome (DS) caused by a trisomy of chromosome 21 (HSA21), is the most common genetic developmental disorder, with an incidence of one in 800 live births. DS individuals show cognitive impairment, learning and memory deficits, arrest of neurogenesis and synaptogenesis, and early onset of Alzheimer's disease (AD; Antonarakis et al, 2004; Lott & Dierssen, 2010). The detailed pathogenetic mechanisms by which the extra copy of HSA21 leads to the neurodevelopmental DS phenotype remain unknown. High-resolution mapping of rare, partial trisomies of HSA21 has provided evidence that several regions exist on HSA21 with various ‘dosage sensitive’ genes contributing to a given phenotype, which could also be modified by other genes on HSA21 and in the rest of the genome (Lyle et al, 2008; Korbel et al, 2009).
Several approaches have been used to study the pathogenesis of DS. Mouse aneuploidies have been engineered to model the various DS phenotypes; these models display some behavioral, anatomical and cellular abnormalities similar to human phenotypes (reviewed in Das & Reeves, 2011). Additionally, studies with post-mortem human DS brains have allowed the investigation of neuroanatomical abnormalities and the alterations that occur during brain development (Becker et al, 1991; Mito & Becker, 1993; Guidi et al, 2008). Moreover, in vitro culture of neural progenitor cells (NPCs) isolated from brain of DS patients were informative in dissecting the mechanisms underlying brain defects (Bahn et al, 2002; Esposito et al, 2008; Bhattacharyya et al, 2009; Lu et al, 2011). The discovery that pluripotent stem cells (PSCs) can be induced from human adult somatic cells by the introduction of few factors and their extensive similarities to embryonic stem cells (ESCs; Takahashi et al, 2007; Yu et al, 2009) has provided new opportunities for investigation in human developmental biology and diseases. Recent studies have been successful in generating disease-specific induced pluripotent stem cells (iPSCs) from a variety of neurodevelopmental and neurodegenerative disorders (Park et al, 2008; Soldner et al, 2009; Brennand et al, 2011; Israel et al, 2012), providing excellent in vitro models for the pathophysiology and potential treatment of these disorders (Hibaoui & Feki, 2012).
In the present study, we have generated iPSCs from fetal fibroblasts of monozygotic twins discordant for trisomy 21: Twin-N-iPSCs for the normal iPSCs and Twin-DS-iPSCs for the iPSCs carrying the trisomy 21. The use of monozygotic twins has allowed us to study the effect of the supernumerary chromosome 21 without the biological ‘noise’ of the variation of the genome. We evaluated their multi-lineage potentials in vivo by teratoma formation when iPSCs were injected intramuscularly into immunodeficient SCID mice. The DS pathogenesis was further investigated when iPSCs were induced to differentiate into neural progenitor cells (NPCs) and neurons. Furthermore, we used Twin-DS-iPSCs to validate candidate genes involved in the impairment of neurogenesis described in DS patients. Among the numerous protein coding genes of HSA21, dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A ( DYRK1A) encodes a proline-directed serine/threonine and tyrosine kinase which plays pleiotropic roles in neurodevelopment and disease (Tejedor et al, 1995; Guimera et al, 1996; Song et al, 1996; Becker et al, 1998; Tejedor & Hämmerle, 2011). Transgenic mice carrying an extra copy of Dyrk1A display neurodevelopmental delays, motor abnormalities, learning and memory deficits, and altered synaptic plasticity (Smith et al, 1997; Altafaj et al, 2001; Ahn et al, 2006). Based on these studies, we investigated the role of DYRK1A in Twin-DS-iPSC-derived NPCs and neurons, through its inhibition by pharmacological means or by short hairpin RNA silencing.
Results
Generation of iPSCs from monozygotic twins discordant for trisomy 21
Normal (Twin-N) and Down syndrome (Twin-DS) fetal fibroblasts were isolated from monozygotic twins discordant for trisomy 21 (Dahoun et al, 2008) and used to establish Twin-N-iPSCs and Twin-DS-iPSCs using OCT4, SOX2, KLF4 and c-MYC genes as previously described (Takahashi et al, 2007; Grad et al, 2011; Fig 1A and B). All iPSC lines expressed markers of pluripotent cells including NANOG, OCT4, TRA-1-60, TRA-1-81 and SSEA4 as demonstrated by immunofluorescence staining (Fig 1C) and showed alkaline phosphatase activity (supplementary Fig S1A). Quantitative (qRT-PCR) and non quantitative RT-PCR analysis demonstrated expression of selected endogenous pluripotent transcription factors including OCT4, SOX2, NANOG, LIN28 and ZFP42 ( REX1) in the generated iPSCs in contrast with the parental fibroblasts (Fig 1D and supplementary Fig S1B). As expected for cells that have acquired a pluripotent state, transgene silencing was verified after initial expansion of around 10 passages of Twin-N-iPSCs and Twin-DS-iPSCs (supplementary Fig S1B). Moreover, as in hESC-H1, OCT4 and NANOG promoter regions of Twin-N-iPSCs and Twin-DS-iPSCs were found hypomethylated in comparison with their parental fibroblasts (Fig 1E).
Figure 1.

- Schematic representation of Twin-N and Twin-DS parental fibroblasts reprogramming into Twin-N-iPSCs and Twin-DS-iPSCs using OCT4, SOX2, KLF4 and c-MYC genes
- Phase contrast images of Twin-N-iPSCs and Twin-DS-iPSCs growing on feeder cells.
- Immunofluorescence staining of Twin-N-iPSC and Twin-DS-iPSC lines for pluripotency markers NANOG, OCT4, SSEA4, TRA1-60 and TRA1-80.
- qRT-PCR of pluripotency-related genes; NANOG, OCT4, SOX2, LIN28 and ZFP42 ( REX1). Data are represented as mean ± s.e.m. from n = 3.
- DNA methylation profile of OCT4 and NANOG promoters. The global percentage of methylated cytosines (% Me) is indicated (open and closed circles indicate unmethylated and methylated CpGs, respectively).
Transcriptome dysregulation of DS iPSCs
These iPSCs were evaluated to confirm the disease-specific genotype of their parental somatic cells. As revealed by karyotype and array-based comparative genomic hybridization analysis, Twin-DS-iPSCs showed the characteristic trisomy 21 while Twin-N-iPSCs had a normal karyotype (Fig 2A and B). Then, whole transcriptome analysis using mRNA-Sequencing confirmed that the majority of HSA21 genes are indeed more expressed in the trisomic lines than the euploid lines (Fig 2C and supplementary Fig S2A) which is consistent with the overall up-regulation of HSA21 genes in individuals with DS (Antonarakis et al, 2004; Lott & Dierssen, 2010). Principal component analysis (PCA) showed that the first principal component (PC1) clearly discriminated the trisomic from the euploid samples, explaining most of the variance in the whole genome data (72%) and in the analysis of HSA21 genes (91%; Fig 2D and supplementary Fig S2B).
Figure 2.
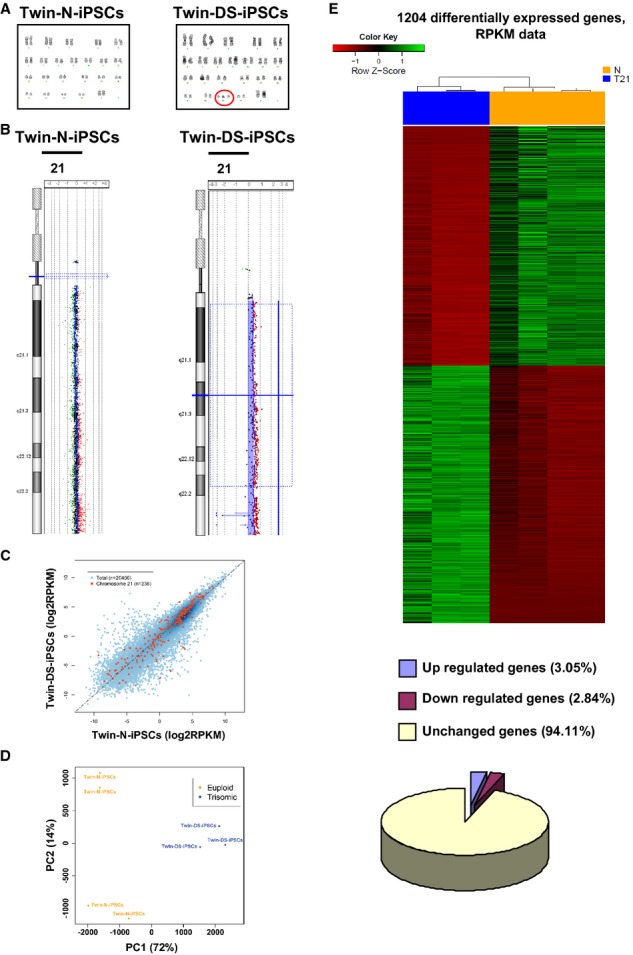
- Karyotypes of Twin-N-iPSCs and Twin-DS-iPSCs are 46, XX and 47, XX+21, respectively.
- Ideogram and array-CGH profile showing the gain of one DNA copy of chromosome 21 in Twin-DS-iPSCs in comparison with Twin-N-iPSCs.
- Comparison of the normalized gene expression levels mean of log2 RPKM between Twin-N-iPSCs and Twin-DS-iPSCs (HSA21 genes are shown in red and the rest of the genome in blue).
- Principal component analysis (PCA) plot based on the normalized expression values in Twin-N-iPSCs and Twin-DS-iPSCs. The percentages represent the proportion of variance explained by each component.
- Heat map of the normalized gene expression values in Twin-DS-iPSCs and Twin N-iPSCs for the 1204 differentially expressed genes. A negative z-score (in red) indicates low expression (below the mean) whereas a positive z-score (in green) shows high expression (above the mean). Diagram showing the proportion of genes differentially expressed between Twin-N-iPSCs and Twin-DS-iPSCs. See also supplementary Table S2.
Differential expression analysis was performed using the statistical R packages EdgeR and DESeq. We considered the genes reported as being differentially expressed by both packages thereby obtaining a conservative list of 580 downregulated and 624 upregulated genes expressed in Twin-DS-iPSCs (Bonferroni-corrected P-value < 0.01; Fig 2E and supplementary Table S2). To further characterize the biological processes that might be affected in Twin-DS-iPSCs, we investigated the functional annotations associated with the 1204 differentially expressed genes using DAVID (Database for Annotation, Visualization and Integrated Discovery). The gene ontology (GO) analysis of the 580 genes downregulated in Twin-DS-iPSCs revealed significant enrichment for genes involved in multiple developmental processes including embryonic development and morphogenesis, organ development and morphogenesis and system development. Among these genes, 96 genes were associated with nervous system-related terms and specifically with nervous system development, central and peripheral nervous system development, brain development, neurogenesis, generation of neurons, neuron differentiation and axonogenesis (Table 1 and supplementary Fig S4). The GO analysis also revealed enrichment for terms linked to cellular adhesion (i.e. biological adhesion, cell adhesion, cell-cell adhesion) and to the cadherin signalling pathway. Additionally, WNT signalling and cancer pathways were found associated with the list of downregulated genes. The GO analysis of the 624 genes upregulated in Twin-DS-iPSCs showed enrichment for functions related to the regulation of RNA metabolic processes, regulation of transcription and DNA-dependent transcription. Interestingly, an important number of zinc finger protein genes were upregulated in Twin-DS-iPSCs (Table 1 and supplementary Table S3).
Table 1.
Gene ontology (GO) categories of biological processes in which significant differentially expressed genes were grouped
| Category | Term | Count | % | P-value | Benjamini |
|---|---|---|---|---|---|
| GO categories of biological processes asssociated with the 580 downregulated genes: | |||||
| GO:0007275 | Multicellular organismal development | 189 | 33.6 | 1.10E-29 | 2.40E-26 |
| GO:0048731 | System development | 165 | 29.3 | 1.60E-28 | 1.70E-25 |
| GO:0032502 | Developmental process | 195 | 34.6 | 3.20E-27 | 2.30E-24 |
| GO:0048856 | Anatomical structure development | 170 | 30.2 | 6.20E-27 | 3.40E-24 |
| GO:0009653 | Anatomical structure morphogenesis | 103 | 18.3 | 8.20E-23 | 3.60E-20 |
| GO:0007399 | Nervous system development | 95 | 16.9 | 2.30E-21 | 8.20E-19 |
| GO:0048513 | Organ development | 125 | 22.2 | 3.40E-21 | 1.00E-18 |
| GO:0032501 | Multicellular organismal process | 222 | 39.4 | 3.40E-21 | 9.30E-19 |
| GO:0030154 | Cell differentiation | 117 | 20.8 | 1.70E-19 | 4.20E-17 |
| GO:0048869 | Cellular developmental process | 118 | 21 | 1.50E-18 | 3.30E-16 |
| GO:0007156 | Homophilic cell adhesion | 30 | 5.3 | 1.80E-17 | 3.60E-15 |
| GO:0022610 | Biological adhesion | 68 | 12.1 | 1.90E-17 | 3.40E-15 |
| GO:0007155 | Cell adhesion | 67 | 11.9 | 6.60E-17 | 1.90E-14 |
| GO:0016337 | Cell-cell adhesion | 41 | 7.3 | 9.70E-17 | 1.70E-14 |
| GO:0009887 | Organ morphogenesis | 54 | 9.6 | 1.50E-13 | 2.20E-11 |
| GO:0007389 | Pattern specification process | 36 | 6.4 | 2.20E-13 | 3.00E-11 |
| GO:0009888 | Tissue development | 59 | 10.5 | 2.40E-13 | 3.10E-11 |
| GO:0001501 | Skeletal system development | 39 | 6.9 | 4.20E-13 | 5.10E-11 |
| GO:0009790 | Embryonic development | 53 | 9.4 | 7.50E-13 | 8.50E-11 |
| GO:0048598 | Embryonic morphogenesis | 37 | 6.6 | 2.90E-12 | 3.10E-10 |
| GO:0022008 | Neurogenesis | 52 | 9.2 | 2.20E-11 | 2.30E-09 |
| GO:0043009 | Chordate embryonic development | 37 | 6.6 | 2.60E-11 | 2.60E-09 |
| GO:0009792 | Embryonic development ending in birth or egg hatching | 37 | 6.6 | 3.40E-11 | 3.20E-09 |
| GO:0048699 | Generation of neurons | 48 | 8.5 | 1.90E-10 | 1.80E-08 |
| GO:0006357 | Regulation of transcription from RNA polymerase II promoter | 56 | 9.9 | 2.70E-10 | 2.40E-08 |
| GO:0050793 | Regulation of developmental process | 52 | 9.2 | 1.30E-09 | 1.10E-07 |
| GO:0051239 | Regulation of multicellular organismal process | 63 | 11.2 | 3.80E-09 | 3.10E-07 |
| GO:0030182 | Neuron differentiation | 39 | 6.9 | 5.10E-09 | 3.90E-07 |
| GO categories of biological processes asssociated with the 624 upregulated genes: | |||||
| GO:0051252 | Regulation of RNA metabolic process | 106 | 2.1 | 2.40E-09 | 5.60E-06 |
| GO:0006355 | Regulation of transcription, DNA-dependent | 104 | 2.1 | 3.10E-09 | 3.60E-06 |
| GO:0006350 | Transcription | 117 | 2.4 | 4.60E-09 | 3.60E-06 |
| GO:0009058 | Biosynthetic process | 169 | 3.4 | 4.20E-08 | 2.40E-05 |
| GO:0044249 | Cellular biosynthetic process | 165 | 3.3 | 5.00E-08 | 2.30E-05 |
| GO:0009059 | Macromolecule biosynthetic process | 140 | 2.8 | 1.60E-07 | 6.30E-05 |
| GO:0045449 | Regulation of transcription | 131 | 2.6 | 1.80E-07 | 5.90E-05 |
| GO:0034645 | Cellular macromolecule biosynthetic process | 139 | 2.8 | 1.80E-07 | 5.30E-05 |
| GO:0051171 | Regulation of nitrogen compound metabolic process | 139 | 2.8 | 3.30E-07 | 8.60E-05 |
| GO:0019219 | Regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 138 | 2.8 | 3.50E-07 | 8.00E-05 |
The table lists the biological processes that might be affected due to trisomy 21, GO categories are ranked by P-values and Benjamini-corrected P-values given by DAVID (see also supplementary Table S3). The number of genes differentially expressed in each category is also shown.
In vivo and in vitro differentiation of normal and DS iPSCs
To document their developmental potential in vivo, iPSCs were injected intramuscularly into immunodeficient SCID mice. Histological analysis revealed that Twin-N-iPSCs formed teratoma with all embryonic germ layers: mesoderm, ectoderm and endoderm (Fig 3A upper panel and supplementary Fig S4). In contrast, injection of Twin-DS-iPSCs formed teratoma with multiple cysts which were surrounded by an abundant undifferentiated mesenchyme (Fig 3A lower panel). Mucinous intestinal-type differentiation or foci of bone were also found (Fig 3A lower panel). Surprisingly, differentiated ectoderm germ structures were not found in Twin-DS-iPSC-derived teratomas (Fig 3A lower panel). These results were confirmed by the near absence of staining for the neuroepithelial marker NESTIN in Twin-DS-iPSC-derived teratoma (supplementary Fig S4). We next determined that these iPSCs could differentiate in vitro into all three embryonic germ layers as detected by expression of the ectodermal marker β3-TUBULIN, the mesodermal marker α-SMOOTH MUSCLE ACTIN (α-SMA) and the endodermal marker α-FETOPROTEIN (AFP; Fig 3B).
Figure 3.
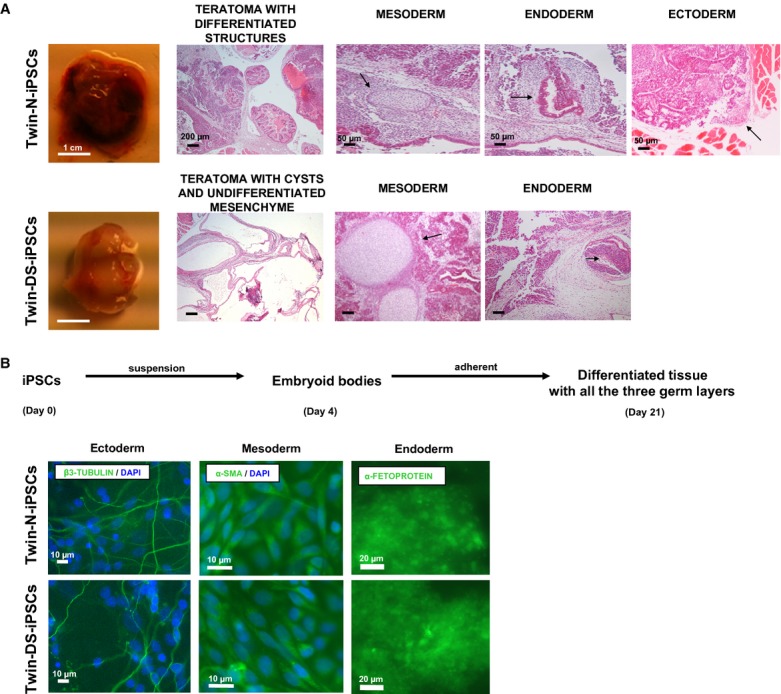
- Hematoxylin and eosin staining analysis of teratoma generated after intramuscular injection of Twin-N-iPSCs (upper panel) and Twin-DS-iPSCs (lower panel) into SCID mice. See also supplementary Fig S4.
- Spontaneous in vitro differentiation of Twin-N-iPSCs (upper panel) and Twin-DS-iPSCs (lower panel) as embryoid bodies (EBs) in suspension culture for 4 days and as adherent cells for an additional 17 days. These EBs expressed α-SMA (mesoderm), AFP (endoderm) and β3-tubulin (ectoderm).
Proliferation deficit and increased apoptosis in NPCs derived from DS iPSCs
IPSC lines were also characterized to confirm their differentiation potentials into neural progenitor cells (NPCs, day 21) in vitro through the protocol outlined in Fig 4A. As expected, the expression of the pluripotency markers OCT4 and NANOG decreased in both iPSCs when induced to differentiate into NPCs (Fig 4B). Under these conditions, we next investigated the kinetics of emergence of neuronal and non-neuronal markers in these cells. Interestingly, our results showed that NPCs derived from Twin-DS-iPSCs expressed more AFP than those derived from Twin-N-iPSCs (Fig 4C). In contrast, we did not find any difference in the expression of the endodermal marker GATA4 and the mesodermal markers SMA and T (Fig 4C and D). Moreover, the expression of the neuroepithelial precursor marker NES and the neuronal markers TUBB3, MAP2 and FOXA2 were lower in NPCs derived from Twin-DS-iPSCs than those derived from Twin-N-iPSCs (Fig 4E). In addition, Twin-DS-iPSC-derived cells exhibited a more astroglial phenotype as revealed by the greater expression of GFAP, S100B and VIM (Fig 4F). Also, we detected an increased expression of the basic helix-loop-helix transcription factors and oligodendroglial markers OLIG1 and OLIG2 in Twin-DS-iPSC-derived NPCs (Fig 4F).
Figure 4.

A Schematic representation of neural induction of the iPSCs into NPCs and neuronal differentiation into neurons.
B–F qRT-PCR analysis of pluripotency, endodermal and mesodermal markers upon neural induction of Twin-N-iPSCs and Twin-DS-iPSCs into NPCs. The expression of neuronal, astroglial and oligodendroglial markers is also shown. Data are represented as mean ± s.e.m. For clarity, statistics are only shown at the NPC level (day 21). Ns non significant, * P < 0.05, ** P < 0.01 by Student's t-test from n = 3–5.
G Proportion of SOX2/NESTIN double positive cells in NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs. See also supplementary Fig S5.
H Cell proliferation analysis by Ki-67 staining of NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs. See also supplementary Fig S6.
I Nuclear damage analysis of NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs (arrows, Hoechst 33342 staining).
J Representative AMC fluorescence traces and quantification of caspase-3 activity in NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs. Data are represented as mean ± s.e.m. *** P < 0.001 by Student's t-test from n > 4. See also supplementary Fig S7.
To test whether the neurogenesis defect is associated with a decrease of NPC population in DS neurospheres, we analyzed the number of NESTIN and SOX2 double positive cells in dissociated neurospheres derived from Twin-N-iPSCs and Twin-DS-iPSCs. Importantly, we found a significant decrease of NESTIN/SOX2 double positive cells in Twin-DS-iPSC-derived NPCs (88.9 ± 3.8 versus 56.4 ± 2.9%, P < 0.0001 for neurospheres derived from Twin-N-iPSCs and Twin-DS-iPSCs, respectively; Fig 4G), indicating a reduced number of NPCs in Twin-DS-iPSC-derived neurospheres. Further analysis revealed an increased proportion of committed astroglial cells (2.4 ± 0.6 versus 13.0 ± 1.6%, P = 0.0009 for GFAP+ cells in neurospheres derived from Twin-N-iPSCs and Twin-DS-iPSCs, respectively) and oligodendroglial cells (3.7 ± 2.0 versus 11.7 ± 0.9%, P = 0.0055 for OLIG2+ cells in neurospheres derived from Twin-N-iPSCs and Twin-DS-iPSCs, respectively) in neurospheres derived from Twin-DS-iPSCs (supplementary Fig S5B). The proportion of committed neuronal cells had a tendency to be higher in neurospheres derived from Twin-DS-iPSCs but failed to reach significance (5.6 ± 0.9 versus 14.7 ± 3.6%, P = 0.089 for β3-TUBULIN+ cells in neurospheres derived from Twin-N-iPSCs and Twin-DS-iPSCs, respectively; supplementary Fig S5B). Next, we assessed whether the defective neurogenesis in DS is due to proliferation deficits and/or increased cell death of NPCs. As shown in Fig 3H, the proportion of Ki-67 positive cells is significantly lower in Twin-DS-iPSC-derived NPCs consistent with a reduced proliferation of Twin-DS-iPSC-derived NPCs. Moreover, NPCs derived from Twin-DS-iPSCs showed an approximately threefold increase of the number of apoptotic nuclei together with an approximately twofold increase of caspase-3 activity compared with those derived from Twin-N-iPSCs (Fig 4I and J). This proliferation deficit and increased apoptosis affected principally NESTIN positive cells as revealed by an approximately twofold decrease of the number of cells double positive for NESTIN and Ki-67 and an approximately four-and six-fold increase of the number of cells double positive for NESTIN and cleaved caspase-3 in neurospheres derived from Twin-DS-iPSCs (supplementary Figs S6 and S7).
Altered maturation of NPCs derived from DS iPSCs into neurons
When NPCs were further induced to mature into neurons, we found a decreased expression of β3-TUBULIN and MAP2, and an increase of GFAP, VIMENTIN, S100B, OLIG1 and OLIG2 markers in Twin-DS-iPSC-derived cells when compared with Twin-N-iPSC-derived cells, as revealed by immunofluorescence staining and qRT-PCR (Fig 5A and B). These results strongly support an impaired neurogenesis together with a shift from neuronal to astroglial and oligodendroglial phenotype in Twin-DS-iPSC-derived cells. Notably, this shift was still observed even when a greater number of NPCs was induced to mature into neurons which rules out the possibility that the reduced number of NPCs derived from Twin-DS-iPSCs could lead to the observed shift from neuronal to astroglial and oligodendroglial phenotype (unpublished observations). Quantification of neurite (either axons or dendrites) branching from soma of β3-TUBULIN positive neurons showed that the mean number of branches was approximately twofold lower in neurons derived from Twin-DS-iPSCs than those derived from Twin-N-iPSCs (Fig 5C). Thus, neurons derived from Twin-DS-iPSCs demonstrated reduced length of neurites (Fig 5D).
Figure 5.

A Quantitative expression of neuronal (β3-TUBULIN and MAP2), astroglial (GFAP) and oligodendroglial (OLIG2) markers after maturation of NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs into neurons, by immunofluorescence analysis. The proportion of β3-TUBULIN, GFAP and OLIG2 positive cells is also shown. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01, *** P < 0.001 by Student's t-test from n > 4.
B qRT-PCR analysis of neuronal ( TUBB3, MAP2 and FOXA2), astroglial and oligodendroglial ( GFAP, S100B, VIM, OLIG1 and OLIG2) markers upon neuronal differentiation of NPCs into neurons. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01, *** P < 0.001 by Student's t-test from n = 4.
C Representative images and quantitative analysis of neurites (either axons or dendrites) from the soma of β3-TUBULIN positive neurons derived from Twin-N-iPSCs and Twin-DS-iPSCs. Data are represented as mean ± s.e.m. *** P < 0.001 by Student's t-test from n = 4.
D Quantitative analysis of the length of neurites of neurons derived from Twin-N-iPSCs and Twin-DS-iPSCs. Data are represented as mean ± s.e.m. *** P < 0.001 by Student's t-test from n = 4.
E, F Quantitative analysis of MAP2 positive neurons derived from Twin-N-iPSCs and Twin-DS-iPSCs stained for SYNAPSIN1 (in E), PSD95 and GAD67 (in F). Data are represented as mean ± s.e.m. Ns non significant, * P < 0.05, ** P < 0.01 by Student's t-test from n = 3–4.
G qRT-PCR of the synaptic markers SYN1, PSD95, SNPA25 and GAD67 in neurons derived from Twin-N-iPSCs and Twin-DS-iPSCs. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01, *** P < 0.001 by Student's t-test from n = 3–5.
An impaired synaptic maturation together with an imbalance between excitatory and inhibitory synapses have been proposed to explain the cognitive impairment in DS (Kleschevnikov et al, 2004; Belichenko et al, 2007; Chakrabarti et al, 2007; Martínez-Cué et al, 2013). Therefore, we investigated the expression of both pre-and post-synaptic markers in neurons by immunofluorescence staining and qRT-PCR analysis. MAP2-positive dendrites in neurons derived from Twin-DS-iPSCs exhibited a reduced density of SYNAPSIN punctae (Fig 5E) and a reduced expression of SYN1 and SNAP25 transcripts (Fig 5G). We next investigated the proportion of GABA-ergic and glutamatergic neurons derived from Twin-N-iPSCs and Twin-DS-iPSCs. Interestingly, we found a reduced density of PSD95, a post-synaptic protein expressed in glutamatergic neurons and a lower expression of PSD95 transcripts in neurons derived from Twin-DS-iPSCs (Fig 5F and G). In contrast, we found a greater expression of GAD67 (which encodes an enzyme responsible for the synthesis of GABA in neurons) in Twin-DS-iPSC-derived neurons (Fig 5G). The expression of GAD67 protein had a tendency to be higher in neurons derived from Twin-DS-iPSCs but failed to reach significance (Fig 5F).
Rescue of neurogenesis impairment of NPCs and neurons derived from DS iPSCs through DYRK1A inhibition
Considering that DYRK1A is involved in neurodevelopment and altered in DS (Tejedor et al, 1995; Guimera et al, 1996; Song et al, 1996; Tejedor & Hämmerle, 2011), we investigated the role of DYRK1A in NPCs and neurons derived from Twin-DS-iPSCs. As shown in Fig 6A, an approximately twofold increase of DYRK1A protein was found in Twin-DS-iPSC-derived NPCs (see also supplementary Fig S8). Moreover, treatment of Twin-DS-iPSCs during neural induction into NPCs with the selective DYRK1A inhibitor epigallocatechine gallate (EGCG, 10 μM) resulted in a significant inhibition of DYRK1A activity which reached almost the value of control (Fig 6B and C). Therefore, we tested whether DYRK1A inhibition by EGCG treatment during neural induction and neuronal differentiation of Twin-DS-iPSCs, can affect the defective neurogenesis of these cells. Notably, EGCG treatment improved the number of NPCs derived from Twin-DS-iPSCs by promoting proliferation and preventing apoptosis (Fig 6D–F). To provide additional support for the role of DYRK1A in these defects and to exclude a possible antioxidant effect of EGCG, knockdown of DYRK1A was achieved using short hairpin RNA (shRNA) in Twin-DS-iPSCs (Fig 6G–I and supplementary Fig S6). Importantly, this knockdown improved the number of NPCs derived from Twin-DS-iPSCs by promoting proliferation and preventing apoptosis (Fig 6J–L). When these NPCs were further induced to mature into neurons, DYRK1A inhibition through EGCG treatment or shRNA improved the expression of the neuronal markers β3-TUBULIN and MAP2, indicative of an improvement of neurogenesis (Fig 7A–D).
Figure 6.

A Expression and activity of DYRK1A in NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs. Data are represented as mean ± s.e.m. ** P < 0.01 by Student's t-test from n = 4.
B Schematic representation for generation of NPCs from Twin-DS-iPSCs after incubation with EGCG 10 μM.
C Activity of DYRK1A in NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs. The effect of 10 μM EGCG is also shown. Data are represented as mean ± s.e.m. ** P < 0.01, *** P < 0.001 by one-way ANOVA followed with Tukey's test from n = 5.
D–F Effect of DYRK1A inhibition by EGCG on the number (in D), the proliferation (in E, Ki-67 staining) and cell death (in F, caspase-3 activity) of NPCs derived from Twin-DS-iPSCs. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01 by one-way ANOVA followed with Tukey's test from n = 4.
G Schematic representation for generation of NPCs from Twin-DS-iPSCs after transduction with lentiviruses encoding shRNAs targeting DYRK1A.
H–L Expression (in H) and activity (in I) of DYRK1A in NPCs derived from Twin-N-iPSCs, Twin-DS-iPSCs and Twin-DS-iPSCs with DYRK1A shRNA. Effect of DYRK1A inhibition by shRNA on the number (in J), the proliferation (in K, Ki-67 staining) and cell death (in L, caspase-3 activity) of NPCs derived from Twin-DS-iPSCs. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01, *** P < 0.001 by one-way ANOVA followed with Tukey's test from n = 4.
Figure 7.
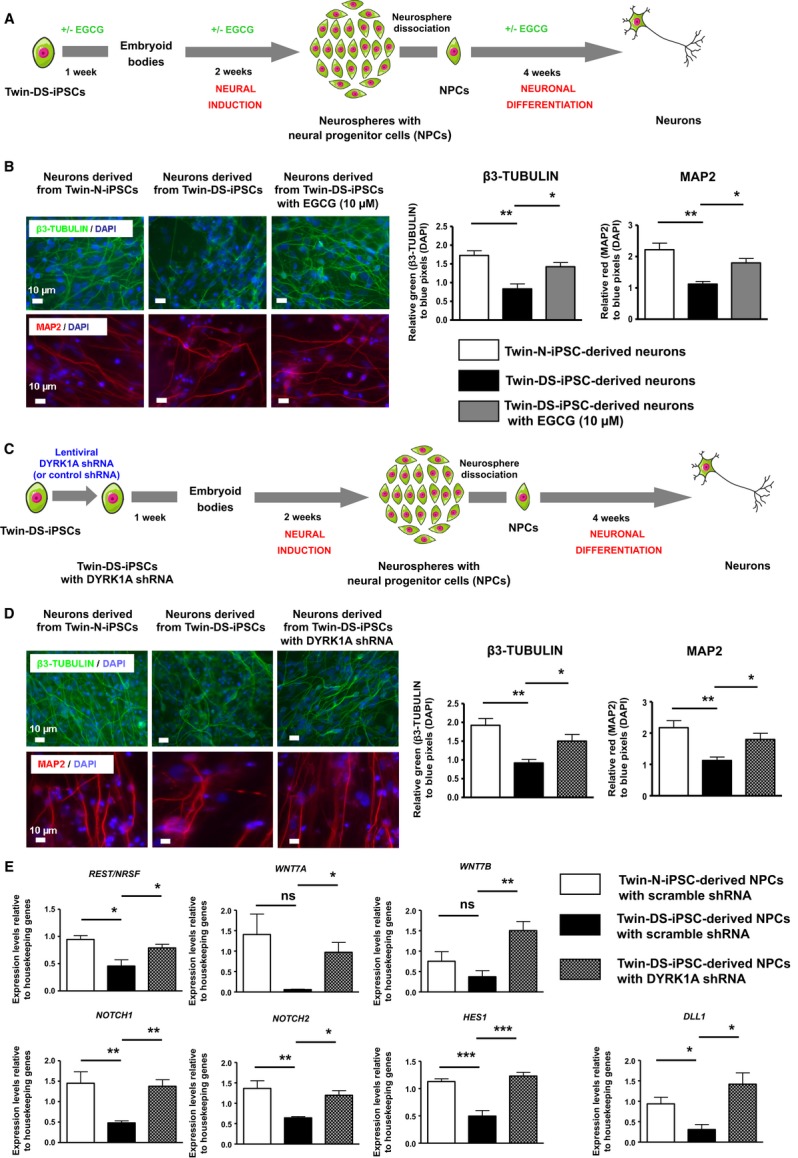
- Schematic representation for generation of neurons from Twin-DS-iPSCs after incubation with EGCG 10 μM.
- Effect of DYRK1A inhibition by EGCG on the expression of neuronal markers upon neuronal differentiation of Twin-DS-iPSCs. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01 by one-way ANOVA followed with Tukey's test from n = 4.
- Schematic representation for generation of neurons from Twin-DS-iPSCs after transduction with lentiviruses encoding shRNAs targeting DYRK1A.
- Effect of DYRK1A inhibition through shRNA silencing on the expression of neuronal markers upon neuronal differentiation of Twin-DS-iPSCs. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01 by one-way ANOVA followed with Tukey's test from n = 4.
- qRT-PCR of REST/NRSF, WNT7A, WNT7B, NOTCH1, NOTCH2, HES1 and DLL1 in NPCs derived from Twin-N-iPSCs and Twin-DS-iPSCs. The effect of DYRK1A inhibition by shRNA is also shown. Data are represented as mean ± s.e.m. * P < 0.05, ** P < 0.01, *** P < 0.001 by one-way followed with Tukey's test from n = 4–7.
To explore the possible mechanisms underlying these improvements, we further investigated the expression of crucial genes and pathways involved in neural fate and differentiation namely REST/NRSF (Kuwabara et al, 2004; Ballas et al, 2005), WNT and NOTCH signalling pathways (Ciani & Salinas, 2005; Ables et al, 2011). Consistent with a previous report (Canzonetta et al, 2008), qRT-PCR clearly showed that REST/NRSF expression was reduced by approximately 30% in Twin-DS-iPSC-derived NPCs in comparison with Twin-N-iPSC-derived NPCs (Fig 7E). Moreover, we investigated the expression of NOTCH1 and NOTCH2 receptors, the NOTCH ligand DLL1 and the NOTCH target HES1. As shown in Fig 7E, NPCs derived from Twin-DS-iPSCs exhibited a 2.9-and 2.1-fold reduction of NOTCH1 and NOTCH2 respectively, as well as an approximately twofold reduction of DLL1 and HES1, in comparison with those derived from Twin-N-iPSCs. Similarly, the near absence of expression of WNT7A in NPCs derived from Twin-DS-iPSCs resulted in a 28-fold reduction of it expression compared to those derived from Twin-N-iPSCs (Fig 7E). Thus, WNT7B was also found downregulated in these cells (Fig 7E). Importantly, restoring DYRK1A expression in Twin-DS-iPSC-derived NPCs to near normal levels by shRNA, promoted the expression of these genes (Fig 7E).
Discussion
A major challenge in the field of DS research has been to recapitulate the disease features and to understand the molecular mechanisms by which the extra copy of HSA21 genes leads to the abnormalities observed in DS patients. Many attempts have been made to identify HSA21 genes involved in the cognitive impairment in DS patients. For instance, several studies reported genotype-phenotype correlations in DS using genetic analysis (Lyle et al, 2008; Korbel et al, 2009); however, the molecular mechanisms underlying most of the DS phenotypes remain unknown. In order to recapitulate the disease phenotype in vitro, both ESCs and iPSCs have been derived from embryos and patients with DS (Park et al, 2008; Biancotti et al, 2010; Shi et al, 2012). Shi et al reported a PSC model of AD pathology using DS iPSCs. Neurons derived from DS iPSCs exhibited greater secretion of amyloid peptides, tau protein phosphorylation and cell death, supporting the notion that DS iPSCs are an excellent model for AD study (Shi et al, 2012). To our best knowledge, our study is the first demonstrating a neurodevelopmental phenotype in human iPSC-derived neural population. Twin-N-iPSCs and Twin-DS-iPSCs were derived from monozygotic twins discordant for trisomy 21 and thus confounding effects from genomic variability were theoretically eliminated. Twin monozygosity and discordance for trisomy 21 anomaly were tested by cytogenetic, genotyping and BAC-array analysis in the case-report published by Dahoun and colleagues (Dahoun et al, 2008). Since the rest of the genome is identical between the two samples, we hypothesized that these twins were ideal to study the effect of the supernumerary HSA21.
Several observations and findings arise from the current study investigating the cellular and molecular characteristics of neural cells derived from Twin-DS-iPSCs. In this regard, transcriptional profiling has proven extremely informative for the study of many diseases (Brennand et al, 2011; Israel et al, 2012). First, the genetic profiling of Twin-DS-iPSCs confirms that trisomy 21 not only affected the expression of trisomic genes but also the expression of disomic genes in Twin-DS-iPSCs, which is consistent with the argument that trisomy 21 leads to the alteration of whole transcriptome (Lyle et al, 2008). Among these genes, we identified 96 downregulated genes related to brain-related functions (supplementary Fig S4). Then, network analysis further implicates several expected and new pathways involving developmental processes (including those related to neurogenesis), cancer pathways, regulation of transcription, zinc finger (Znf)-C2H2 type genes and cadherins (Fig 2F, Table S3 and supplementary Fig S9). Collectively, the observation of these alterations in Twin-DS-iPSCs illustrates the transcriptional signature of the early developmental abnormalities of DS.
Another major finding of this study is that Twin-DS-iPSCs demonstrated an abnormal in vivo and in vitro neural differentiation. In particular, the teratoma formation assay revealed an abnormal in vivo differentiation of Twin-DS-iPSCs into neurectodermal tissue. In line with this, Mensah et al (2007) showed that the introduction of HSA21 into a transchromosomic mouse ESC line caused the suppression of in vivo differentiation into neurectoderm in a teratoma formation assay. Moreover, by tracking the neural induction and differentiation of iPSCs, it was possible to identify a phenotypic disturbance during early neural development of Twin-DS-iPSCs. In comparison with neurospheres derived from Twin-N-iPSCs, those derived from Twin-DS-iPSCs showed a reduced number of NPCs that was associated with a proliferation deficit and an increased apoptosis. These results are consistent with the increased rate of apoptosis and decreased proliferation capacity observed in brains from DS patients and mouse models of DS (Contestabile et al, 2007; Guidi et al, 2008; Lu et al, 2011).
Furthermore NPCs derived from Twin-DS-iPSCs revealed a decreased expression of neuronal markers and an increased expression of glial markers. In this respect, numerous morphometric studies have shown a lower neuronal density in several parts of human brains including the hippocampal dendate gyrus, the hippocampus, the parahippocampal gyrus and the cerebellum (Guidi et al, 2008, 2011). Our results are further supported by studies showing an increased ratio of astrocytes/neurons in human DS brain (Mito & Becker, 1993; Guidi et al, 2008). In NPCs isolated from brain of DS fetuses, there is evidence both in favor and against the idea that trisomy 21 favors glial at the expense of neuronal differentiation. These discrepancies may be attributable to the brain region origin, the different gestational ages of the fetal tissue and the methodologies used for isolation, maintenance and differentiation of these NPCs. For instance, Bahn and colleagues have shown a reduced number of neurons differentiated from fetal DS NPCs but revealed no difference in the number of astroglial cells (Bahn et al, 2002). In another study, fetal DS NPCs showed no alterations at early stages of neural development but only at later stages. In particular, these NPCs generated fewer neurons and showed increased expression of oligodendroglial markers when they were expanded for more than 10 weeks (but unchanged when expanded for <6 weeks; Bhattacharyya et al, 2009). In contrast, other studies demonstrated that even at early stages of neural development, fetal DS NPCs are less neurogenic and adopt a more gliocentric progenitor phenotype as revealed by the reduced expression of neuroepithelial and neuronal markers ( NESTIN, PAX6, MAP2 and DCX) together with the increased expression of several glial markers ( GFAP, S100B, OLIG1, OLIG2, CNPase, O4 and PDGFRA; Esposito et al, 2008; Lu et al, 2011, 2012). Considering the key roles of S100B, GFAP and VIM in glial differentiation (Lu et al, 2011), the overexpression of these astroglial genes in NPCs derived from Twin-DS-iPSCs leads ultimately to more gliogenic progenitors and the generation of astroglial cells upon differentiation. Moreover, the oligodendroglial shift induced by the overexpression of OLIG1 and OLIG2 in Twin-DS-iPSC-derived cells is in accordance with the pivotal role of these HSA21 genes in specifying oligodendrocyte lineage in the central nervous system of vertebrate (Zhou & Anderson, 2002). Collectively, our results suggest that NPCs derived from Twin-DS-iPSCs fail to generate the same number of neurons as those derived from Twin-N-iPSCs due to defects in NPCs rather than in survival of the generated neurons after 4 weeks of differentiation. It is important to note, however, that we cannot exclude that neurons derived from Twin-DS-iPSCs could be more susceptible to apoptosis after long-term culture, reflecting in some extent late occurring neurodegeneration observed in DS patients (Shi et al, 2012).
Neurons derived from Twin-DS-iPSCs exhibited not only a reduction of their population but also structural alterations compared to those derived from Twin-N-iPSCs. We found in particular a reduced number and length of neurites from soma of neurons derived from Twin-DS-iPSCs which is consistent with previous morphometric studies on brain of DS patient (Becker et al, 1991) and in neurons differentiated from fetal DS NPCs (Bahn et al, 2002). In line with this, we present evidence that neurons derived from Twin-DS-iPSCs exhibit a synaptic phenotype similar to that observed in DS patients and in mouse models of DS (Becker et al, 1991; Dierssen et al, 2003; Chakrabarti et al, 2007). Neurons derived from Twin-DS-iPSCs exhibit a reduced density of SYNAPSIN punctae. Considering the key role of the pre-synaptic proteins such as SYNAPSIN, SNAP25 in the regulation of transmitter release, their reduced expression may alter synaptic function in neurons derived from Twin-DS-iPSCs. Moreover, excess inhibitory synapses at the expense of excitatory ones has been proposed to explain the abnormal synaptic plasticity in mouse models of DS (Kleschevnikov et al, 2004; Belichenko et al, 2007; Chakrabarti et al, 2007; Martínez-Cué et al, 2013). In the present study, we found a reduced expression of PSD95 in neurons derived from Twin-DS-iPSCs, indicative of a lower proportion of excitatory glutamatergic synapses. In addition, the proportion of inhibitory GABA-ergic synapses stained positive for GAD67 was not substantially altered in neurons derived from Twin-DS-iPSCs. This contrasts with the enhanced immunoreactivity of proteins associated with GABA-ergic synapses reported in several brain regions of mouse model of DS (Belichenko et al, 2007; Mazur-Kolecka et al, 2012; Martínez-Cué et al, 2013). Although a recent study showed no difference in the excitatory post-synaptic properties of neurons derived from normal and DS iPSCs (Shi et al, 2012), it will be interesting in the future to investigate the excitatory and inhibitory post-synaptic properties of neurons derived from our iPSCs.
Given that HSA21 contains hundreds of genes, it has been a challenge to identify critical genes responsible for the cognitive impairment in DS patients. Among the candidate genes, DYRK1A has received increased interest due to its implication in neurodevelopment in flies, mice and humans (Tejedor et al, 1995; Guimera et al, 1996; Song et al, 1996; Guedj et al, 2009; Tejedor & Hämmerle, 2011; Mazur-Kolecka et al, 2012). Both loss and gain of function of DYRK1A result in neurodevelopmental defects. Dyrk1A−/− null mutant mice show growth delay and die during midgestation whereas mice heterozygous for Dyrk1A mutation ( Dyrk1A−/+) display a brain size 30% smaller than wild type with a region-specific reduction of neurons (Fotaki et al, 2002; Benavides-Piccione et al, 2005). In humans, DYRK1A haploinsufficiency is associated with microcephaly, growth and mental retardation (Møller et al, 2008; Yamamoto et al, 2011). Moreover, Dyrk1A transgenic mice carrying an extra copy of Dyrk1A, exhibit features similar to those observed in DS patients including hippocampal-dependent memory tasks, arrest of neurogenesis and altered synaptic plasticity (Smith et al, 1997; Altafaj et al, 2001; Ahn et al, 2006; Das & Reeves, 2011; Martinez de Lagran et al, 2012). The present study provides novel evidence that DYRK1A overexpression is a main contributor to the impaired neurogenesis observed in NPCs and neurons derived from Twin-DS-iPSCs. This was strongly supported by the findings that DYRK1A inhibition by EGCG or shRNA improved proliferation of NPCs derived from Twin-DS-iPSCs. Several additional lines of evidence reinforced the involvement of DYRK1A in the control of cell growth and neurogenesis. DYRK1A has been shown to phosphorylate p53 which leads to the up-regulation of p53 target genes associated with cell cycle regulation and more particularly p21CIP1 in rat and human NPCs as well as in brains from Dyrk1A transgenic mice and DS patients. The induction of p21CIP1 impairs G1/G0-S phase transition, inhibiting NPC proliferation (Park et al, 2010). In another study, Yabut et al demonstrated that Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of NPCs (Yabut et al, 2010). They showed that Dyrk1A overexpression through in utero electroporation inhibits neural cell proliferation in the embryonic neocortex. This effect was mediated by the nuclear export and degradation of cyclin D1, a cyclin required for cell proliferation by allowing the entry to the S phase (Yabut et al, 2010). More recently, Hämmerle and colleagues showed that Dyrk1A promotes cell cycle exit through induction of the expression of the cyclin-dependent kinase inhibitor p27KIP1 in neural precursors, which further binds to and inhibits the cyclin/cyclin-dependent kinase complex that controls G1/S transition (Hämmerle et al, 2011). Moreover, Litovchick and colleagues demonstrated that DYRK1A plays an important role in suppression of mammalian cell proliferation. In particular, DYRK1A overexpression promotes quiescence and senescence through DREAM (DP, RB, E2F and MuvB) complex assembly, a complex involved in cell cycle processes (Litovchick et al, 2011).
Contrary to the numerous reported roles of DYRK1A in the control of cell cycle, very little is known regarding its impact on cell death. For instance, DYRK1A has been shown to prevent the intrinsic apoptotic pathway through the phosphorylation of caspase-9 during retina development (Laguna et al, 2008). In contrast, the overexpression of Dyrk1A makes rat embryonic hippocampal progenitor cells more susceptible to apoptosis by phosphorylating and activating p53 which leads to the subsequent upregulation of p53 target genes and proteins such as FAS (Park et al, 2010). This is of special interest as the protein levels of the pro-apoptotic genes p53 and FAS are increased in the cerebral cortex and the cerebellum of DS patients (de la Monte et al, 1998; Seidl et al, 1999). In the present study, we provide novel evidence that the increased expression and activity of DYRK1A is responsible for the greater susceptibility to apoptosis of NPCs derived from Twin-DS-iPSCs. Reducing DYRK1A expression and activity to near control values prevented the apoptosis of these cells. It remains to be established whether p53 and FAS are responsible for the increase in apoptosis induced by DYRK1A overexpression in NPCs derived from Twin-DS-iPSCs.
Another important observation is that DYRK1A inhibition by EGCG treatment or by shRNA increased the number of neurons derived from Twin-DS-iPSCs. Consistent with a previous study (Canzonetta et al, 2008), this effect was associated with an increase of the expression of REST/NRSF, a transcription factor that activates genes encoding fundamental functions during neuronal differentiation including neural lineage specification, synapse formation and function (Kuwabara et al, 2004; Ballas et al, 2005; Abrajano et al, 2009). In this regard, the downregulation of REST/NRSF in fetal DS NPCs has been shown to lead to the subsequent downregulation of important regulators of cell adhesion and synaptic plasticity (Bahn et al, 2002). In agreement with this report, we found a reduced expression of REST/NRSF in NPCs and of SYN1 in neurons derived from Twin-DS-iPSCs. Moreover, recent studies point to an important role of DYRK1A in dendrites tree development, synapse formation and function (Tejedor & Hämmerle, 2011; Martinez de Lagran et al, 2012). For instance, single overexpression of Dyrk1A in vivo is sufficient to recapitulate the dendritic alterations observed in DS patients (Martinez de Lagran et al, 2012). In line with this, we found that DYRK1A inhibition by EGCG or by shRNA promotes the dendritic development of neurons derived from Twin-DS-iPSCs. This effect was associated with a restoration of the expression to near normal levels of WNT7A, WNT7B, NOTCH1, NOTCH2, DLL1 and HES1. This is of special interest as these genes are crucial regulators of dendrite morphogenesis and synapse assembly (Ciani & Salinas, 2005; Breunig et al, 2007; Ables et al, 2011) and could explain at least in part, the defects in dendritic development due to DYRK1A overexpression in the present study and recent reports (Guedj et al, 2009; Tejedor & Hämmerle, 2011; Martinez de Lagran et al, 2012). Of note, when NPCs derived from Twin-DS-iPSCs were treated with EGCG during neuronal differentiation only (and not before, during neural induction), no improvement in the number of neurons was found. However, this delayed targeting of DYRK1A by EGCG promoted dendritic development and the density of the synaptic protein SYNAPSIN in these neurons (supplementary Fig S10). These results further highlight the pleiotropic roles of DYRK1A during the sequential stages of neural induction and neuronal differentiation of iPSCs as well as the potential opportunities for DS therapy through DYRK1A targeting in early and late neurodevelopmental events.
In conclusion, the generation of iPSCs from monozygotic twins discordant for trisomy 21 provides a unique model to study the effects of trisomy 21 of two otherwise genetically identical samples (Dahoun et al, 2008). This model recapitulates neurodevelopmental features of DS and provides important clues to the physiopathology of the disease (supplementary Fig S11). Our study further emphasizes DYRK1A as a main contributor of neurogenesis impairment in NPCs and neurons derived from Twin-DS-iPSCs and provides novel insights into the therapeutic potential of DYRK1A targeting in patients with DS. It is important to note, however, that other genes including OLIG, NOTCH and WNT deserve further investigation. Finally, the finding that DYRK1A inhibition by pharmacological and genetic approaches contributes to reverse the abnormal neurogenesis in NPCs and neurons derived from Twin-DS-iPSCs, allows a proof-of-principle for potential screening tests using iPSC technology (Hibaoui & Feki, 2012) and should provide the basis for designing new therapeutic approaches for patients with DS.
Materials and Methods
Cell culture and iPSC derivation
Primary fetal skin fibroblasts were isolated from monozygotic twins discordant for trisomy 21 (Twin-N for the normal fibroblasts and Twin-DS for the trisomy 21) and used to establish normal (Twin-N-iPSCs) and Down syndrome (Twin-DS-iPSCs) induced pluripotent stem cells (iPSCs). These samples were obtained post mortem and the study was approved by the ethics committee of the Geneva University Hospital. These iPSCs were generated by transducing the parental fibroblasts (Twin-N and Twin-DS) with polycistronic lentiviral vectors expressing OCT4, SOX2, KLF4 and c-MYC genes (Takahashi et al, 2007; Grad et al, 2011). Colonies that appeared were isolated and expanded into lines (see also supplementary Table S1). H1-hESCs (obtained from WiCell Research Institute, Madison, WI, USA) and the generated iPSCs were cultured on irradiated human foreskin fibroblasts (ATCC; CCD 1112Sk, Manassas, VA, USA) that were mitotically inactivated by irradiation at 35 Gy before seeding on a gelatin-coated 6-well plate at 3.5 × 105 cells/plate. IPSC colonies were maintained with daily changes in Knock-out Dulbecco's Minimal Essential Medium (KO DMEM) supplemented with 20% KO serum replacement, 1 mM l-glutamine, 100 μM non-essential amino acids, 100 μM 2-mercaptoethanol, 50 U/ml penicillin and 50 mg/ml streptomycin (all from Gibco, Invitrogen, Basel, Switzerland) and 100 ng/ml human basic fibroblast growth factor (bFGF; Peprotech, London, UK). All iPSC lines were passaged by manual dissection of cell clusters in the presence of 10 μM ROCK-inhibitor Y-27632 (Sigma-Aldrich, Buhs, Switzerland).
Spontaneous in vitro three-germ layer differentiation
Whole iPSC colonies were seeded into ultra low attachment dishes (Costar; Corning Life Sciences, Schiphol-Rijk, The Netherlands) in KO DMEM medium supplemented with 10% new calf serum, 1 mM l-glutamine, 100 μM non-essential amino acids, 100 μM 2-mercaptoethanol, 50 U/ml penicillin and 50 mg/ml streptomycin. Within 24 h, cells aggregated to form embryoid bodies (EBs). After 4 days growing in suspension, these EBs were transferred to gelatin-coated dishes containing the same medium to allow the cells to differentiate for an additional 17 days.
Neural induction of iPSCs into NPCs and neuronal differentiation into neurons
Differentiation of iPSCs into neural progenitor cells (NPCs) and mature neurons was established as described with minor modifications (Suter et al, 2009; Brennand et al, 2011). Briefly, the colonies were mechanically detached from the feeder cells, washed and maintained in suspension in ultra-low attachment dishes (Costar, Corning Life Sciences) for 1 week in neural induction medium (NIM) consisting of DMEM-F12, 1% penicillin/streptomycin, N2 supplement (Gibco, Invitrogen) and replaced with NIM supplemented with 10 ng/ml human recombinant bFGF for another 2 weeks. Thereafter, NPCs were further induced to mature into neurons by plating the dissociated aggregates on poly-ornithine/laminin-coated tissue culture plate in neural differentiation medium (NDM) consisting of neurobasal supplemented with B-27 supplement (Gibco, Invitrogen), 10 ng/ml BDNF (R&D Systems, Inc., Minneapolis, MN, USA), 10 ng/ml GDNF (R&D Systems, Inc.), 1 mM dibutyryl-cyclic AMP, 200 nM ascorbic acid (Sigma-Aldrich, Buhs) and 1% penicillin/streptomycin for 4 weeks. Medium was changed every 3 days.
Teratoma formation from iPSCs
All the procedures involving animals were conducted in accordance with the Swiss Federal Veterinary Office's guidelines, based on the Swiss Federal Law on Animal Welfare. In vivo differentiation studies were carried out by teratoma formation. 5 × 106 cells from each iPSCs were harvested and injected intramuscularly into SCID mice. After 8 weeks, the teratomas were excised, fixed in 4% formaldehyde, and embedded in paraffin for hematoxylin and eosin (HE) staining or for immunohistochemistry analysis with horseradish peroxidase system. 4 μm thick teratomas sections from formalin fixed paraffin embedded samples were analyzed by immunohistochemistry using anti-alpha-fetoprotein (NCL-AFPp, Novocastra Laboratories Ltd, Newcastle upon Tyne, UK), anti-alpha-smooth muscle actin (M0851, Dako, Baar, Switzerland) and anti-nestin antibodies (MAB1259, R&D systems, Inc.) with the Ventana Discovery automated staining system (Ventana Medical Systems, Tucson, AZ, USA). Ventana reagents for the entire procedure were used. For both AML and AFP antibodies, no antigen retrieval pretreatment was required. After automatic deparaffinization, slides were incubated 30 min at 37°C with primary antibodies respectively diluted at 1/300 (AML) and 1/750 (AFP) in antibody diluent from Dako (S2022). For nestin antigen retrieval, slides were heated in CC1 cell conditioning solution for 36 min (EDTA antigen retrieval solution pH 8.4) using a standard protocol. Anti-nestin antibodies were used at dilution 1/1000 and also incubated 30 min at 37°C. Detection of primary antibodies was carried out using the secondary universal biotinylated antibodies reagent and the amplified DABMap kit (Ventana Medical Systems), based on conversion of diaminobenzidine to a dye with multimeric horseradish peroxidase (HRP). As negative control, AFP antibodies were replaced by rabbit control immunoglobulins and AML and nestin antibodies were replaced by mouse IgG serotype control.
Immunocytochemistry
Briefly, iPSCs at different stages of neuronal differentiation were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 30 min, permeabilized with 0.2% Triton X-100 for 30 min, and blocked with 5% bovine serum albumin in PBS for 1 h at room temperature (RT). Cells were incubated with primary antibody overnight at 4°C, washed with PBS and incubated with secondary antibody for 1 h at RT. All secondary antibodies were tested for crossreactivity and nonspecific immunoreactivity. The antibodies used for immunohistochemical staining are listed in supplementary Table S4. The cells were finally mounted in UltraCruz ™ mounting medium containing DAPI for nuclei identification (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Images of the immunostained cells were captured on a Zeiss Axioskop 2 fluorescence microscope with an Axiocam Color HRc detector (Zeiss, Feldbach, Switzerland). Staining of cells derived from Twin-N-iPSCs and Twin-DS-iPSCs, neurite length and number were analyzed with ImageJ software. N values for the immunocytochemical analyses represent at least four independent experiments in triplicate.
Karyotype analysis
Karyotyping was performed on at least twenty metaphase spreads using the CTG-banding method. IPSCs at 80% confluence were treated with 0.2 μg/ml colcemid (Invitrogen) for 2 h and harvested. Cell pellets were resuspended in pre-warmed hypotonic solution (KCl, 0.075 mol/l) for 10 min at 37°C. Cells were then fixed with freshly prepared, ice-cold methanol-acetic acid solution (3:1 in volume) and mounted by dropping onto slides.
Bisulfite sequencing analysis
About 2 μg of DNA extracted from the iPSCs and the parental fibroblasts were bisulfite-converted and purified using Epitect Bisulfite Kits (Qiagen, Valencia CA, USA) following the manufacturer's instruction. DNA bisulphite treatment and processing were performed simultaneously for all cell lines. The promoter regions of OCT4 and NANOG were amplified with specific primers previously described (Freberg et al, 2007) using JumpStart REDTaq DNA Polymerase (Sigma-Aldrich, Buhs) and KAPA2G Robust DNA Polymerase (Kapabiosystems, Boston, MA, USA). Quantitative methylation percentages were determined by Epitect sequencing (QIAGEN GmbH, Hilden, Germany). The Epitect sequencing service includes sequencing of the PCR products using cycle sequencing (Reactions were cycled in a GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA, USA) and purified using DyeEx™ (QIAGEN). Data collection was carried out on a 3730xl DNA Analyzer (Applied Biosystems). It also includes modified analysis of raw data to obtain a percentage of methylation for each CpG dinucleotide. The methylation percentages of these sites were averaged to obtain a single methylation score for each sample.
Array CGH
Array-Comparative Genomic Hybridization (CGH) was performed using the Human Genome CGH Microarray Kit 44A (Agilent Technologies, Palo Alto, CA, USA) with a resolution of about 150 kb. Briefly, 1 μg of iPSC DNAs were processed according to the manufacturer's protocol. Fluorescence was scanned in a dual-laser scanner (Agilent DNA microarray scanner G2565CA; Agilent Technologies) and the images were extracted and analysed with Agilent Feature Extraction software (v9.5.3.1) and Workbench analysis software (v7.0) respectively. Changes in test DNA copy number at a specific locus are observed as the deviation of the log ratio value from a modal value of 0. The data files have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE52251 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52251).
RNA extraction, non-quantitative and quantitative real time polymerase chain reaction
Total RNA was extracted from the cell lines, using the QIAGEN RNeasy MiniKit according to the manufacturer's protocol (Invitrogen). RNA integrity and quantity were assessed with an Agilent 2100 bioanalyser (Agilent Technologies, Santa Clara, CA, USA), using RNA 6000 nanochips. 1 μg of total RNAs were reverse transcribed with the Superscript III reverse transcriptase (Invitrogen) according to the manufacturer's protocol. One-twentieth cDNA template was used as template for each PCR reaction. cDNA was real time polymerase chain (PCR) amplified in a 7900HT Sequence Detection Systems (Applied Biosystems) using the Power SYBR Green PCR master mix (Applied Biosystems). Raw threshold-cycle ( Ct) values were obtained with the Sequence Detection Systems 2.0 software (Applied Biosystems). Melting curve analysis were automatically performed to monitor production of the appropriate PCR product. Relative quantities (RQs) were calculated with the formula RQ = E− Ct, using efficiencies calculated for each run with the Data Analysis for Real-Time PCR (DART-PCR) algorithm, as described (Peirson et al, 2003). A mean quantity was calculated from triplicate PCR reactions for each sample, and this quantity was normalized against that of the housekeeping genes ( GusB and eEF1). Each PCR reaction was performed at least in triplicate with negative controls and the mean quantities were calculated from them. For non-quantitative PCR, reactions were performed in a Biometra thermocycler (Göttingen, Germany), with RedTaq polymerase mix (Sigma-Aldrich, St. Louis, MO, USA), 250 nM primers and 1 μl of cDNA. The primer sequences used for quantitative and non quantitative RT-PCR are listed in supplementary Table S4.
Gene expression analysis by mRNA sequencing
mRNA-Seq libraries were prepared from 500 ng of total RNA using the Illumina TruSeq™ RNA Sample Preparation kit (S-930-2001, Illumina, Inc., San Diego, CA, USA), following the manufacturer's instructions. Libraries were sequenced on the Illumina HiSeq 2000 instrument (Illumina, Inc.) to generate 100 bp paired-end reads. Those reads were mapped against the genome (hg 19) using the default parameters of the Burrows-Wheeler Aligner (BWA; Li & Durbin, 2009). An expression signal was detected in at least one sample for 20'456 genes (comprising also non-coding RNA). For each gene, the level of expression was determined by calculating the exon coverage (custom pipeline) and normalizing in Reads per Kilobase per Million (RPKM). Twin-DS-iPSC and Twin-N-iPSC samples were sequenced in three and four biological replicates, respectively, starting from different RNA preparations. Principal component analyses (PCA) and clustering analyses were performed using R (version 2.14.0, http://www.R-project.org/). Differential expression between the trisomic and the normal replicates was assessed using the default parameters of EdgeR (version 2.4.6; Robinson et al, 2010) and DESeq (version 1.6.1; Anders & Huber, 2010) programs in R (version 2.14.0). Only the genes with more than 1 read per million in at least three replicates were conserved for this analysis. Bonferroni correction was applied to adjust for multiple testing. A gene was considered differentially expressed if the Bonferroni-corrected P-value was lower than 0.01 with both methods. Analysis of the functional annotations associated with the differentially expressed genes was performed using Database for Annotation, Visualization and Integrated Discovery (Huang et al, 2008, 2009). REVIGO interactive graph of the top biological processes was performed as previously described (Supek et al, 2011). The data files have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE52251 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52251).
Cell death assays
Caspase-3 activity was determined in NPC homogenates as previously described (Hibaoui et al, 2009). Caspase–3 activity was measured by monitoring the proteolytic cleavage of the fluorogenic substrate N–acetyl-Asp-Glu-Val-Asp-7–amino–4–methylcoumarin (Ac-DEVD–AMC; Biomol, Anawa Trading, Zürich, Switzerland) for 30 min at 30°C using a FLUOstar OPTIMA fluorimeter (BMG Labtech, Champigny sur Marne, France; excitation 380 nm, emission 460 nm). The caspase–3 inhibitor N–acetyl-Asp-Glu-Val-Asp-aldehyde (Ac–DEVD–CHO; Biomol) was used at 10 μM as a control. Caspase-3 activity was expressed as AUF per minute 100 μg per protein (AUF for arbitrary units of fluorescence). Protein content was determined using the Bradford method (BioRad, Reinach, Switzerland). Nuclear morphology of NPCs was studied by using the cell permeable DNA-binding dye Hoechst 33342 (Invitrogen). NPCs with homogeneously stained, regularly and roundly shaped nuclei were considered to be normal NPCs whereas NPCs that exhibited reduced nuclear size, chromatin condensation, intense fluorescence, and nuclear fragmentation were considered as apoptotic. NPCs were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde and stained with 1 μg/ml of Hoechst 33342 for 5 min at room temperature. Nuclear morphological changes were visualized using Zeiss Axioskop 2 fluorescence microscope with an Axiocam Color HRc detector (Zeiss). Nuclei from several randomly chosen areas were counted and the number of apoptotic nuclei was expressed as percentage of the total number of nuclei.
DYRK1A kinase assay
NPCs were lysed by up-and-down pipetting with a lysis buffer (50 mM Tris-HCl pH7.5, 150 mM NaCl, 0.5% NP-40, 15% glycerol, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 10 μg/ml leupeptin, 1 μl PMSF). After centrifugation (21 000 g, 4°C, 10 min), the supernatant was stored at −80°C. DYRK1A immunoprecipitation was performed using a monoclonal anti-DYRK1A (clone 7D10; Abnova, Taipei City, Taiwan) as previously described with slight modifications (Lilienthal et al, 2010). In vitro DYRK1A kinase activity was measured by incubating the immunoprecipated DYRK1A with the substrate peptide DYRKtide (Catalog#62698; Anaspec, Fremont, CA, USA) in a kinase buffer (25 mM Hepes pH 7.0, 5 mM MgCl2 and 0.5 mM dithiothreitol) containing 40 μM ATP for 3 h at 37°C. DYRK1A kinase activity was measured by quantifying the amount of remaining ATP in the reaction using kinase-Glo luminescent reagent (Promega, Dübendorf, Switzerland). Luminescence was monitored in a plate reader luminometer (Fluostar Optima; BMG Labtechnologies, Offenburg, Germany).
DYRK1A knockdown assay and DYRK1A inhibition with epigallocatechine gallate
Knockdown of DYRK1A was achieved by means of lentiviral vector-mediated short hairpin RNA (shRNA), using validated commercial lentiviral particles for the DYRK1A sequence (Sigma-Aldrich, Buhs). For viral transduction, Twin-DS-iPSCs were plated on matrigel-coated dishes and infected at a multiplicity of infection of 30 in the presence of 10 μg/ml polybrene. As controls, Twin-N-iPSCs and Twin-DS-iPSCs were also infected with control shRNA. Knockdown efficiencies of DYRK1A targeted shRNA were evaluated by real-time RT-PCR and by Western blotting analysis. Moreover, to test the effect of pharmacological DYRK1A inhibition on neurogenesis, Twin-DS-iPSCs were treated with the selective DYRK1A inhibitor epigallocatechine gallate EGCG (Sigma-Aldrich, Buhs) at a concentration of 10 μM in NIM and NDM with medium changes every 2 days. All comparative differentiation experiments were carried out with all experimental lines and controls in parallel.
The paper explained.
Problem
Down syndrome (DS) caused by a trisomy of chromosome 21 (HSA21), is the most common genetic developmental disorder, with an incidence of one in 800 live births. DS individuals show intellectual impairment and several other abnormalities for the majority of which the pathogenetic mechanisms remain unknown. A major challenge has been to recapitulate the disease features and to understand the molecular mechanisms by which the extra copy of HSA21 genes leads to the abnormalities observed in DS patients. Recent studies have been successful in generating disease-specific induced pluripotent stem cells (iPSCs) from a variety of neurodevelopmental and neurodegenerative disorders, providing useful in vitro models.
Results
We report the generation and characterization of iPSCs derived from monozygotic twins discordant for trisomy 21 in order to eliminate the effects of the variability of the genomic background. This provides a unique model to study the effect of the supernumerary HSA21 since the rest of the genome is identical between the two twins. DS iPSCs recapitulate the neurodevelopmental features of DS. First, the alterations observed by genetic analysis at the iPSC level and at first approximation in early development illustrate the developmental disease transcriptional signature of DS. Then, in vivo differentiation of DS iPSCs revealed an abnormal teratoma formation in NOD-SCID mice. In vitro, we found a reduced neurogenesis together with a greater astroglial and oligodendroglial cell population upon neural induction into neural progenitor cells (NPCs) and upon neuronal differentiation of DS iPSCs. This effect was associated with a proliferation deficit and increased apoptosis of NPCs derived from DS iPSCs. In addition, neurons derived from DS iPSCs showed reduced dendritic development and expression of synaptic proteins. Importantly, we provide novel evidence that DYRK1A on HSA21 likely contributes to these defects as its inhibition by pharmacological means or by short hairpin RNA silencing corrected these defects. We found that this correction improved proliferation, decreased apoptosis of NPCs and increased the number of neurons derived from DS iPSCs. Also, DYRK1A correction was associated with the restoration of the expression to near normal levels of crucial regulators of neurogenesis and dendritic development including REST/NRSF, WNT and NOTCH signalling.
Impact
The generation of iPSCs from monozygotic twins discordant for trisomy 21 is an innovative way to study DS neurodevelopment as it offers an unprecedented opportunity to study early embryonic development and enables the investigation of the detailed pathogenetic mechanisms by which the extra copy of HSA21 leads to DS phenotypes. The finding that DYRK1A inhibition by pharmacological and genetic approaches contributes to reversal of the abnormal neurogenesis in NPCs and neurons derived from DS iPSCs, allows a proof-of-principle for potential screening tests using iPSC technology and should provide the basis for designing new therapeutic approaches for DS patients.
Western blotting
NPCs were solubilized at 4°C in 100 μl of lysis buffer and centrifuged to remove debris. Protein were resolved in 7.5% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Incubation with anti-DYRK1A antibody was carried out overnight at 4°C using dilutions of 1:1000 (clone 7D10; Abnova). After incubation with HRP-conjugated secondary antibodies during 1 h, membranes were developed by enhanced chemiluminescence reagents (ECL reagent; Amersham Biosciences) according to the manufacturer's instructions. Signals were quantified and normalized to α-tubulin (clone DM1A T9026; Sigma, Buhs) and expressed as arbitrary units.
Data analysis
The data were analyzed and the graphs were constructed using the GraphPad Prism software (GraphPad, San Diego, CA, USA). Data are shown as mean ± s.e.m. Statistical analysis among groups were evaluated by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test, and comparisons between two groups by Student's t–test. P values of <0.05 were considered significant.
Acknowledgments
This work was supported by grants from the Ernest Boninchi Fondation to A.F., Genico to A.F., the Swiss National Science Foundation (SNF) to S.E.A. and the European Research Council (ERC) to S.E.A. We are grateful to S. Vianin and L. Kocjancic-Curty for their technical support.
Author contributions
YH performed the majority of the experiments and analyzed the data. IG, AL, MRS, SD, MG, and SG performed the experiments. IG, AL, FAS, MFP and FB analyzed data. YH, SEA and AF conceived and designed the study and wrote the manuscript. AF and SEA supervised whole study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for is this article available online: http://www.embomm.embopress.org
References
- Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci. 2011;12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrajano JJ, Qureshi IA, Gokhan S, Zheng D, Bergman A, Mehler MF. REST and CoREST modulate neuronal subtype specification, maturation and maintenance. PLoS One. 2009;4:e7936. doi: 10.1371/journal.pone.0007936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K-J, Jeong HK, Choi H-S, Ryoo S-R, Kim YJ, Goo J-S, Choi S-Y, Han J-S, Ha I, Song W-J. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol Dis. 2006;22:463–472. doi: 10.1016/j.nbd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Altafaj X, Dierssen M, Baamonde C, Martí E, Visa J, Guimerà J, Oset M, González JR, Flórez J, Fillat C, et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum Mol Genet. 2001;10:1915–1923. doi: 10.1093/hmg/10.18.1915. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and Down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5:725–738. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- Bahn S, Mimmack M, Ryan M, Caldwell M, Jauniaux E, Starkey M, Svendsen C, Emson P. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with Down's syndrome: a gene expression study. Lancet. 2002;359:310–315. doi: 10.1016/S0140-6736(02)07497-4. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Becker L, Mito T, Takashima S, Onodera K. Growth and development of the brain in Down syndrome. Prog Clin Biol Res. 1991;373:133–152. [PubMed] [Google Scholar]
- Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost H-G. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem. 1998;273:25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of down syndrome: exploring genotype-phenotype relationships. J Comp Neurol. 2007;504:329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- Benavides-Piccione R, Dierssen M, Ballesteros-Yáñez I, Martínez de Lagrán M, Arbonés ML, Fotaki V, DeFelipe J, Elston GN. Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/− mouse. Neurobiol Dis. 2005;20:115–122. doi: 10.1016/j.nbd.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, McMillan E, Chen SI, Wallace K, Svendsen CN. A critical period in cortical interneuron neurogenesis in Down syndrome revealed by human neural progenitor cells. Dev Neurosci. 2009;31:497–510. doi: 10.1159/000236899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancotti J-C, Narwani K, Buehler N, Mandefro B, Golan-Lev T, Yanuka O, Clark A, Hill D, Benvenisty N, Lavon N. Human embryonic stem cells as models for aneuploid chromosomal syndromes. Stem Cells. 2010;28:1530–1540. doi: 10.1002/stem.483. [DOI] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig JJ, Silbereis J, Vaccarino FM, Šestan N, Rakic P. Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proc Natl Acad Sci USA. 2007;104:20558–20563. doi: 10.1073/pnas.0710156104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzonetta C, Mulligan C, Deutsch S, Ruf S, O'Doherty A, Lyle R, Borel C, Lin-Marq N, Delom F, Groet J, et al. DYRK1A-dosage imbalance perturbs NRSF/REST evels, deregulating pluripotency and embryonic stem cell fate in Down Syndrome. Am J Hum Genet. 2008;83:388–400. doi: 10.1016/j.ajhg.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, Salinas PC. WNTS in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6:351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, Santini D, Bartesaghi R, Ciani E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus. 2007;17:665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- Dahoun S, Gagos S, Gagnebin M, Gehrig C, Burgi C, Simon F, Vieux C, Extermann P, Lyle R, Morris MA, et al. Monozygotic twins discordant for trisomy 21 and maternal 21q inheritance: a complex series of events. Am J Med Genet. 2008;146A:2086–2093. doi: 10.1002/ajmg.a.32431. [DOI] [PubMed] [Google Scholar]
- Das I, Reeves RH. The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis Model Mech. 2011;4:596–606. doi: 10.1242/dmm.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen M, Benavides-Piccione R, Martínez-Cué C, Estivill X, Flórez J, Elston GN, DeFelipe J. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cereb Cortex. 2003;13:758–764. doi: 10.1093/cercor/13.7.758. [DOI] [PubMed] [Google Scholar]
- Esposito G, Imitola J, Lu J, De Filippis D, Scuderi C, Ganesh VS, Folkerth R, Hecht J, Shin S, Iuvone T, et al. Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Hum Mol Genet. 2008;17:440–457. doi: 10.1093/hmg/ddm322. [DOI] [PubMed] [Google Scholar]
- Fotaki V, Dierssen M, Alcántara S, Martínez S, Martí E, Casas C, Visa J, Soriano E, Estivill X, Arbonés ML. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol. 2002;22:6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freberg CT, Dahl JA, Timoskainen S, Collas P. Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell. 2007;18:1543–1553. doi: 10.1091/mbc.E07-01-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad I, Hibaoui Y, Jaconi M, Chicha L, Bergström-Tengzelius R, Sailani MR, Pelte MF, Dahoun S, Mitsiadis TA, Töhönen V, et al. NANOG priming before full reprogramming may generate germ cell tumours. Eur Cell Mater. 2011;22:258–274. doi: 10.22203/ecm.v022a20. [DOI] [PubMed] [Google Scholar]
- Guedj F, Sébrié C, Rivals I, Ledru A, Paly E, Bizot JC, Smith D, Rubin E, Gillet B, Arbones M, et al. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS One. 2009;4:e4606. doi: 10.1371/journal.pone.0004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Bonasoni P, Ceccarelli C, Santini D, Gualtieri F, Ciani E, Bartesaghi R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008;18:180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi S, Ciani E, Bonasoni P, Santini D, Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with Down syndrome. Brain Pathol. 2011;21:361–373. doi: 10.1111/j.1750-3639.2010.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimera J, Casas C, Pucharcôs C, Solans A, Domènech A, Planas AM, Ashley J, Lovett M, Estivill X, Pritchard MA. A human homologue of drosophila minibrain (Mnb) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum Mol Genet. 1996;5:1305–1310. doi: 10.1093/hmg/5.9.1305. [DOI] [PubMed] [Google Scholar]
- Hämmerle B, Ulin E, Guimera J, Becker W, Guillemot F, Tejedor FJ. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development. 2011;138:2543–2554. doi: 10.1242/dev.066167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibaoui Y, Feki A. Human pluripotent stem cells: applications and challenges in neurological diseases. Front Physiol. 2012;3:267. doi: 10.3389/fphys.2012.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibaoui Y, Roulet E, Ruegg UT. Melatonin prevents oxidative stress-mediated mitochondrial permeability transition and death in skeletal muscle cells. J Pineal Res. 2009;47:238–252. doi: 10.1111/j.1600-079X.2009.00707.x. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–20. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Tirosh-Wagner T, Urban AE, Chen X-N, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA. 2009;106:12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell. 2004;116:779–793. doi: 10.1016/s0092-8674(04)00248-x. [DOI] [PubMed] [Google Scholar]
- Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernández E, Fotaki V, Delabar JM, de la Luna S, de la Villa P, Arbonés ML. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev Cell. 2008;15:841–853. doi: 10.1016/j.devcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienthal E, Kolanowski K, Becker W. Development of a sensitive non-radioactive protein kinase assay and its application for detecting DYRK activity in Xenopus laevis oocytes. BMC Biochem. 2010;11:20. doi: 10.1186/1471-2091-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down's syndrome. Lancet Neurol. 2010;9:623–633. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- Lu J, Esposito G, Scuderi C, Steardo L, Delli-Bovi LC, Hecht JL, Dickinson BC, Chang CJ, Mori T, Sheen V. S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS One. 2011;6:e2212. doi: 10.1371/journal.pone.0022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Lian G, Zhou H, Esposito G, Steardo L, Delli-Bovi LC, Hecht JL, Lu QR, Sheen V. OLIG2 over-expression impairs proliferation of human Down syndrome neural progenitors. Hum Mol Genet. 2012;21:2330–2340. doi: 10.1093/hmg/dds052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, Bena F, Gagos S, Gehrig C, Lopez G, Schinzel A, Lespinasse J, Bottani A, Dahoun S, Taine L, et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet. 2008;17:454–466. doi: 10.1038/ejhg.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez de Lagran M, Benavides-Piccione R, Ballesteros-Yañez I, Calvo M, Morales M, Fillat C, DeFelipe J, Ramakers GJA, Dierssen M. Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb Cortex. 2012;22:2867–77. doi: 10.1093/cercor/bhr362. [DOI] [PubMed] [Google Scholar]
- Martínez-Cué C, Martínez P, Rueda N, Vidal R, García S, Vidal V, Corrales A, Montero JA, Pazos Á, Flórez J, et al. Reducing GABAA α5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J Neurosci. 2013;33:3953–3966. doi: 10.1523/JNEUROSCI.1203-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur-Kolecka B, Golabek A, Kida E, Rabe A, Hwang Y-W, Adayev T, Wegiel J, Flory M, Kaczmarski W, Marchi E, et al. Effect of DYRK1A activity inhibition on development of neuronal progenitors isolated from Ts65Dn mice. J Neurosci Res. 2012;90:999–1010. doi: 10.1002/jnr.23007. [DOI] [PubMed] [Google Scholar]
- Mensah A, Mulligan C, Linehan J, Ruf S, O'Doherty A, Grygalewicz B, Shipley J, Groet J, Tybulewicz V, Fisher E, et al. An additional human chromosome 21 causes suppression of neural fate of pluripotent mouse embryonic stem cells in a teratoma model. BMC Dev Biol. 2007;7:131. doi: 10.1186/1471-213X-7-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito T, Becker LE. Developmental changes of S-100 protein and glial fibrillary acidic protein in the brain in Down syndrome. Exp Neurol. 1993;120:170–176. doi: 10.1006/exnr.1993.1052. [DOI] [PubMed] [Google Scholar]
- Møller RS, Kübart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP, Menzel C, Ullmann R, Tommerup N, Ropers H-H, et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet. 2008;82:1165–1170. doi: 10.1016/j.ajhg.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Sohn YK, Ganju N, Wands JR. P53-and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest. 1998;78:401–411. [PubMed] [Google Scholar]
- Park I-H, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Oh Y, Yoo L, Jung M-S, Song W-J, Lee S-H, Seo H, Chung KC. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J Biol Chem. 2010;285:31895–31906. doi: 10.1074/jbc.M110.147520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31:e73. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl R, Fang-Kircher S, Bidmon B, Cairns N, Lubec G. Apoptosis-associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neurosci Lett. 1999;260:9–12. doi: 10.1016/s0304-3940(98)00945-8. [DOI] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Transl Med. 2012;4:124ra129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Stevens ME, Sudanagunta SP, Bronson RT, Makhinson M, Watabe AM, O'Dell TJ, Fung J, Weier HU, Cheng JF, et al. Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat Genet. 1997;16:28–36. doi: 10.1038/ng0597-28. [DOI] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W-J, Sternberg LR, Kasten-Sportès C, Keuren MLV, Chung S-H, Slack AC, Miller DE, Glover TW, Chiang P-W, Lou L, et al. Isolation of human and murine homologues of the drosophila minibrain gene: human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics. 1996;38:331–339. doi: 10.1006/geno.1996.0636. [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter DM, Preynat-Seauve O, Tirefort D, Feki A, Krause KH. Phenazopyridine induces and synchronizes neuronal differentaition of embryonic stem cells. J Cell Mol Med. 2009;13:3517–3527. doi: 10.1111/j.1582-4934.2009.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, Heisenberg M, Fischbach KF, Pongs O. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- Tejedor FJ, Hämmerle B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011;278:223–235. doi: 10.1111/j.1742-4658.2010.07954.x. [DOI] [PubMed] [Google Scholar]
- Yabut O, Domogauer J, D'Arcangelo G. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J Neurosci. 2010;30:4004–4014. doi: 10.1523/JNEUROSCI.4711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Shimojima K, Nishizawa T, Matsuo M, Ito M, Imai K. Clinical manifestations of the deletion of Down syndrome critical region including DYRK1A and KCNJ6. Am J Med Genet A. 2011;155:113–119. doi: 10.1002/ajmg.a.33735. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.