

ABSTRACT
A considerable part of the herpesvirus life cycle takes place in the host nucleus. While much progress has been made to understand the molecular processes required for virus replication in the nucleus, much less is known about the temporal and spatial dynamics of these events. Previous studies have suggested that nuclear capsid motility is directed and dependent on actin filaments (F-actin), possibly using a myosin-based, ATP-dependent mechanism. However, the conclusions from these studies were indirect. They either relied on the effects of F-actin depolymerizing drugs to deduce an F-actin dependency or they visualized nuclear F-actin but failed to show a direct link to capsid motility. Moreover, no direct link between nuclear capsid motility and a molecular motor has been established. In this report, we reinvestigate the involvement of F-actin in nuclear herpesvirus capsid transport. We show for representative members of all three herpesvirus subfamilies that nuclear capsid motility is not dependent on nuclear F-actin and that herpesvirus infection does not induce nuclear F-actin in primary fibroblasts. Moreover, in these cells, three F-actin-inhibiting drugs failed to effect capsid motility. Only latrunculin A treatment stalled nuclear capsids but did so by an unexpected effect: the drug induced actin rods in the nucleus. Immobile capsids accumulated around actin rods, and immunoprecipitation experiments suggested that capsid motility stopped because latrunculin-induced actin rods nonspecifically bind nuclear capsids. Interestingly, capsid motility was unaffected in cells that do not induce actin rods. Based on these data, we conclude that herpesvirus nuclear capsid motility is not dependent on F-actin.
IMPORTANCE
Herpesviruses are large DNA viruses whose replication is dependent on the host nucleus. However, we do not understand how key nuclear processes, including capsid assembly, genome replication, capsid packaging, and nuclear egress, are dynamically connected in space and time. Fluorescence live-cell microscopy revealed that nuclear capsids are highly mobile early in infection. Two studies suggested that this motility might be due to active myosin-based transport of capsids on nuclear F-actin. However, direct evidence for such motor-based transport is lacking. We revisited this phenomenon and found no evidence that nuclear capsid motility depended on F-actin. Our results reopen the question of how nuclear herpesvirus capsids move in the host nucleus.
INTRODUCTION
Cells infected with alphaherpesviruses expressing a fluorescent capsid tag show the remarkable phenotype of high velocity swirling of intranuclear capsids. In 2005, this motion of intranuclear herpes simplex virus 1 (HSV-1) capsids was described as an active, actin-dependent process (1). A year later, we showed that pseudorabies virus (PRV) infection induces intranuclear actin filaments in superior cervical ganglia (SCG) neurons (2). Capsid protein accumulations originally termed assemblons (3)—more likely to be aggregates bearing VP26 fluorescent fusion proteins (4)—were found near these filaments, which also were labeled with anti-myosin V antibody (2). These two reports supported the hypothesis that alphaherpesvirus infection induces intranuclear actin filaments (F-actin) that are the substrates for active capsid motility. This hypothesis remains to be proven rigorously, as the nature and role of filamentous actin in the nucleus are still debated (5–7).
In this report, we reevaluate the conclusions drawn from the results of these two original reports and present evidence that F-actin is not involved in intranuclear capsid motility (NCM) in primary fibroblasts. In previous studies, inhibition of NCM by the F-actin-depolymerizing drug latrunculin A (LatA) was the primary evidence to conclude that NCM depends on F-actin (1). In this report, we visualized F-actin by expressing the live-cell actin marker Lifeact-green fluorescent protein (GFP) or by phalloidin staining after fixation. We demonstrate that F-actin cannot be visualized in the nucleus of infected primary fibroblasts showing NCM. Remarkably, of four F-actin inhibitors tested, only LatA inhibited NCM. We found that concentrations of LatA used in previous reports (1) induced actin rods in some cell types. Actin rods are atypical actin bundles first described 35 years ago in amebae and HeLa cells treated with dimethyl sulfoxide (DMSO) (8). Today, a large body of literature implicates actin rod formation in cellular stress responses to diverse stressors, such as heat shock and ATP depletion. Actin rods also are found in human diseases, such as nemaline myopathy and Alzheimer’s disease. During cellular stress, actin rods seem to sequester actin to prevent F-actin treadmilling (9), reducing the demand on cellular ATP pools. Actin rods form from cofilin-actin subunits assembled into filaments, which then assemble into bigger rod-like structures (10).
We used image correlation microscopy (ICM) to measure nuclear capsid motility. With this technique, we showed that the appearance of LatA-induced nuclear actin rods coincided with the stalling of NCM in cells infected with representative members of all three herpes subfamilies. Inhibiting actin rod formation by cytochalasin D (CytD) also blocked NCM stalling. In addition, we showed that NCM stalling is the result of the direct or indirect binding of the major capsid protein to actin in LatA-treated cells. We concluded that LatA does not depolymerize hypothetical nuclear F-actin, leading to NCM stalling, but instead induces nuclear actin rods that bind capsids and inhibit their motility. Consistent with this hypothesis, capsid motility was not reduced by LatA treatment in a cell line that does not form actin rods.
Our work reopens the question of how nuclear capsids move. It also stresses the complexities induced by drugs that disturb actin homeostasis, particularly LatA, which is often used to investigate the nuclear motility of complexes like herpesvirus replication compartments and cellular chromosomes (11, 12).
RESULTS
Members of all three Herpesvirinae subfamilies show intranuclear capsid motility.
It was previously reported that GFP-tagged human herpesvirus 1 capsids show fast active intranuclear transport that can be inhibited by the F-actin-depolymerizing drug LatA (1). This is an intriguing finding, as the existence of nuclear F-actin and its possible involvement in an active nuclear cargo transport mechanism is a matter of debate (5–7). Understanding how viral capsids move in the host nucleus might help to unveil the basic mechanisms of nuclear F-actin regulation and cargo transport. As a first step, we determined if the phenomenon of nuclear capsid motility is conserved in members of all three Herpesviridae subfamilies.
We used murine embryonic fibroblasts (MEF) because they can be infected by all herpesviruses used in our study. We chose the alphaherpesviruses PRV and HSV-1, the betaherpesvirus murine cytomegalovirus (MCMV), and the gammaherpesvirus murine gammaherpesvirus 68 (MHV-68). For all four viruses, mutants were available in which the small capsid protein (SCP) is tagged with the red fluorescent proteins mRFP or mCherry, allowing nuclear capsids to be visualized in live-cell fluorescence microscopy (13–15) (J. B. Bosse and H. Adler, unpublished data).
Early in infection (6 h postinfection [hpi] for HSV-1, 4 hpi for PRV, 20 hpi for MCMV, and 8 hpi for MHV-68), all four herpesviruses showed nuclear capsid motility (Fig. 1; see also Video S1 in the supplemental material), with nuclei filled with actively moving capsids, while the degree of motility declined at later time points for all four viruses. Capsids from HSV-1 and PRV showed the most motility, and capsids from MCMV and MHV-68 showed less motion (see Video S1).
FIG 1 .
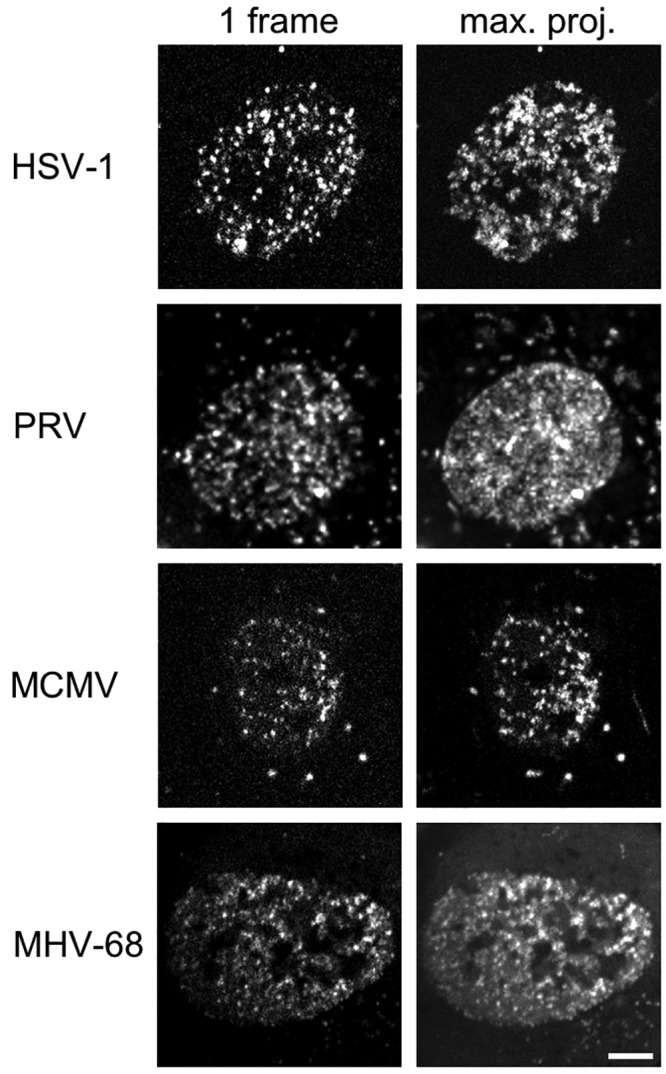
Representative members of all Herpesviridae subfamilies show intranuclear capsid motility. MEF cells constitutively expressing Lifeact-EGFP were infected with HSV-1 mRFP-VP26, PRV mRFP-VP26, MCMV S-mCherry-SCP, or MHV-68 mCherry-ORF65 at a multiplicity of infection (MOI) of at least 5. Live cell spinning disk microscopy with at least 3.7 frames per second was performed in one confocal section after the first capsids appeared in the nucleus at 6 hpi, 4 hpi, 20 hpi, or 8 hpi, respectively. The right column depicts a temporal maximum projection of at least 20 frames representing 5.4 s. The degree of signal smearing indicates the degree of capsid motility. Scale bar indicates 5 µm.
Nuclear actin is not associated with nuclear capsid motion.
We next determined if motility was associated with nuclear F-actin as previously proposed (1, 2). We used MEF cells stably expressing the live cell F-actin probe Lifeact fused to enhanced GFP (EGFP) (MEF-LA) (16, 17). We infected these cells with HSV-1, PRV, MCMV, or MHV-68 to visualize both NCM and Lifeact-labeled actin. We could not detect any nuclear F-actin in cells showing NCM (data not shown).
We also used phalloidin, a well-established F-actin probe. MEF-LA cells were infected, fixed, and stained with fluorescently labeled phalloidin. We used spinning disk microscopy to quantify the amount of nuclear F-actin stained by phalloidin. Out of 102 mock-infected cells, 97 HSV-1-infected cells, 105 PRV-infected cells, 134 MCMV-infected cells, and 167 MHV-68-infected cells, no nuclear F-actin was detected by either phalloidin or Lifeact probes, consistent with the results from live-cell microscopy.
Latrunculin A is the only F-actin-modifying drug that blocks motility of nuclear capsids.
It was previously reported that the F-actin-depolymerizing drug latrunculin A (LatA) stops NCM (1). LatA affects actin polymerization by forming a 1:1 complex with G-actin, which leads to F-actin depolymerization (18). In addition to LatA, we used three different F-actin inhibitors and determined their effect on NCM.
We used image correlation microscopy (ICM) (19) to quantify effects on nuclear capsid motility. ICM is a form of autocorrelation analysis that measures the similarity of an image from one time point to the next in a live-cell movie over time. If significant motility is observed, the similarity between images will decrease quickly, and this is indicated by a drop in the temporal autocorrelation function (TACF). Compared to single-particle tracking, ICM has the advantage of easily integrating the bulk behavior of many capsids without the need to distinguish individual capsids. This fact was particularly important as we used mRFP and mCherry SCP fusions, which produce fewer aggregation artifacts (4) than the EGFP fusions used in previous reports (1, 2). Viruses expressing these fusion proteins also produce more nuclear capsids, making conventional single-particle tracking impossible. ICM is therefore a straightforward way to determine capsid motility in different conditions.
First we analyzed the nuclear capsid motility of HSV-1-, PRV-, MCMV-, and MHV-68-infected MEF-LA cells without actin inhibitors. ICM analysis confirmed the visual impression that HSV-1 and PRV nuclear capsids showed the most motility, while MCMV and MHV-68 capsids showed less motility, as indicated by the faster drop in the TACF (Fig. 2A, black lines).
FIG 2 .
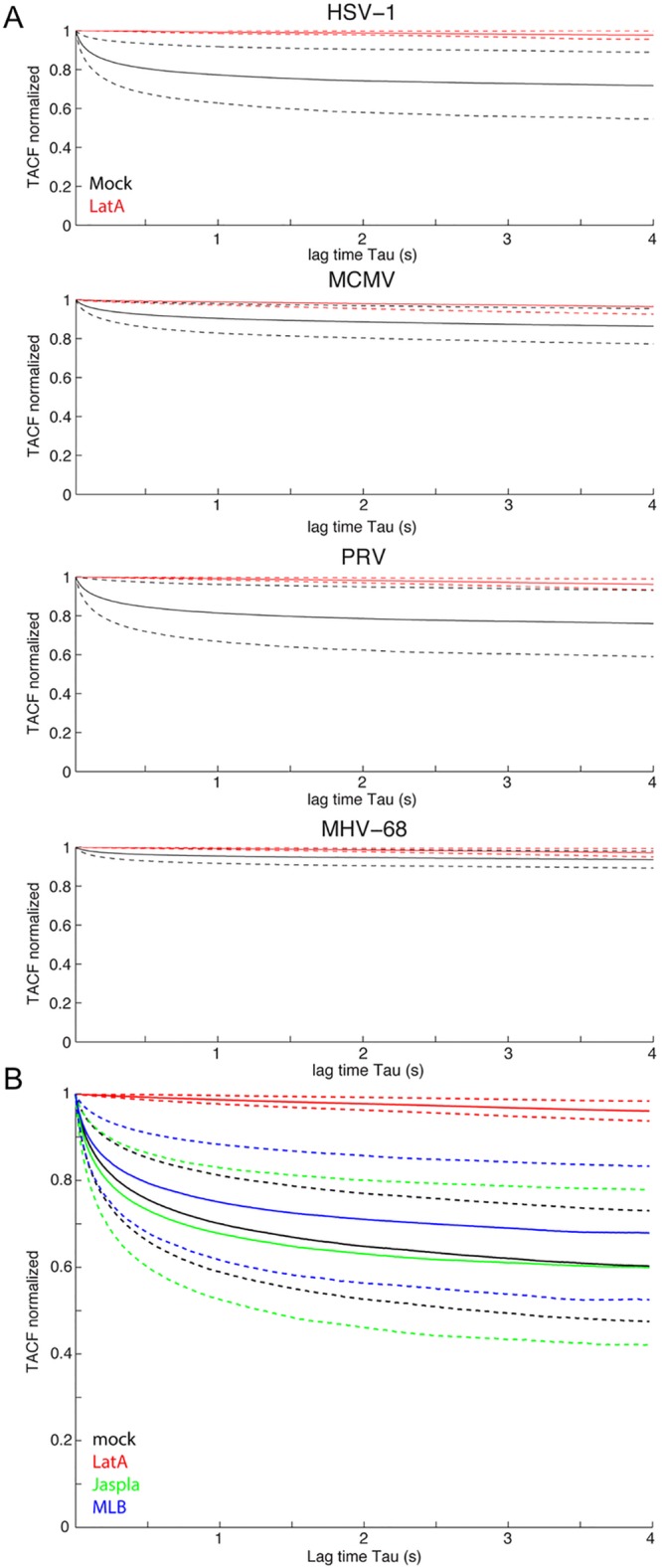
Latrunculin A is the only drug that inhibits nuclear capsid motility. (A) MEF cells constitutively expressing Lifeact-EGFP were infected with HSV-1 mRFP-VP26, PRV mRFP-VP26, MCMV S-mCherry-SCP, or MHV-68 mCherry-ORF65 at an MOI of at least 5. LatA at 4.7 µM (2 µg/ml) (red) was added for 1 h or cells were mock treated (black) at 6 hpi, 4 hpi, 20 hpi, or 8 hpi. (B) MEF cells constitutively expressing Lifeact-EGFP were infected with PRV mRFP-VP26. Cells at 4 hpi were either mock treated (black) or incubated for 1 h with 4.7 µM (2 µg/ml) LatA (red), 1 µM Jaspla (green), or 2 µM MLB (blue). Oblique laser microscopy was performed with 100 frames per second for 10 s, and resulting time-lapse videos of infected nuclei were subjected to autocorrelation analysis. Solid lines indicate mean values; dashed lines indicate the means ± standard deviations. (A) HSV-1, 19 cells mock, 15 cells LatA; PRV, 23 cells mock, 13 cells LatA; MCMV, 21 cells mock, 8 cells LatA; MHV-68, 21 cells mock, 17 cells LatA. (B) Twenty-three cells mock, 14 cells LatA, 29 cells Jaspla, 30 cells MLB. Scale bar indicates 5 µm.
Next, we incubated infected cells 1 h before imaging with 4.7 µM (2 µg/ml) LatA, the concentration used by Forest et al. (1), and performed ICM. LatA treatment inhibited NCM of all four herpesviruses, as indicated by a TACF that remained near 1 (Fig. 2A, red lines).
As all four tested herpesviruses gave similar results, we continued our analyses with PRV. While LatA treatment was effective in inhibiting NCM, it is not clear if the effect is due to direct F-actin disruption. We were especially interested in this possibility of an indirect effect, as it was previously shown that CytD does not have an effect on NCM (1). Therefore, we used two other actin inhibitors with different mechanisms of action. We chose the F-actin depolymerizing drug mycalolide B (MLB), which severs F-actin, binds G-actin in a 1:1 ratio, and has a mechanism of actin depolymerization distinct from the cytochalasins (20). We also analyzed the effects of the F-actin-reorganizing drug jasplakinolide (Jaspla) (1 µM), which leads to unregulated F-actin polymerization and destruction of physiological F-actin structures (21). We determined optimal concentrations of both drugs by testing a wide range of dilutions on MEF-LA cells (see Fig. S1 in the supplemental material). We chose 2 µM of MLB and 1 µM of Jaspla, as these concentrations were the lowest that showed a definite effect on the actin structure. We then measured their effects on capsid motility by ICM. As shown in Fig. 2B, MLB and Jaspla did not have a significant effect on capsid motility compared to that in mock-treated cells. Again, LatA was the only drug that inhibited NCM significantly.
Latrunculin A induces nuclear actin rods which coincide with an inhibition of intranuclear capsid dynamics.
As we could not detect nuclear F-actin, and LatA was the only drug that effected NCM, we speculated that an off-target effect of LatA was responsible for stalling intranuclear capsids. We infected MEF-LA cells with capsid-tagged PRV, treated with LatA, and imaged by fluorescence microscopy. Surprisingly, we observed thick intranuclear structures stained by Lifeact-GFP. Even more surprising, capsids seemed to cluster on and around these structures (Fig. 3A). This was also true for HSV-1, MCMV, and MHV-68 (data not shown). Capsids were mostly immobile, as illustrated by a maximum intensity projection over time (Fig. 3B; see also Video S2 in the supplemental material).
FIG 3 .

Latrunculin A induces nuclear actin rods which coincide with an inhibition of intranuclear capsid dynamics. MEF cells constitutively expressing Lifeact-EGFP were infected with PRV mRFP-VP26. Cells at 4 hpi were either mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA. Cells were fixed and nuclei were stained with DAPI and imaged by confocal laser scanning microscopy (A) or cells were subjected to live-cell spinning disk microscopy (B). A maximum intensity projection of about 1 µm each is shown in the first row of panel A, and a projection of about 8 µm is shown in the second row. (B) One confocal section; the right column represents a temporal maximum projection of 50 frames at 3.7 frames/s. The degree of signal smearing indicates capsid motility. Scale bars indicate 5 µm.
We identified these structures as actin rods, a previously described consequence of a variety of cellular stressors, including LatA treatment (22). A previously postulated (5) explanation for nuclear actin rod formation is that LatA depolymerization of cytoplasmic F-actin may increase the nuclear concentration of G-actin, leading to dysregulated actin polymerization in the nucleus. As DMSO can also induce actin rods, we made sure that the concentrations used to dissolve drugs in this work did not induce actin rods on their own. We found that only DMSO concentrations over 64 µl/ml induced nuclear actin rods in our study system (see Fig. S2 in the supplemental material). We never exceeded 2 µl/ml of DMSO in our final working solutions.
Actin rod induction by LatA was not dependent on Lifeact expression or on virus infection, as normal uninfected MEF cells treated with LatA and stained with phalloidin showed actin rods in the nucleus (see Fig. S3a in the supplemental material). Actin rod formation was dependent on the concentration of LatA used. Reducing the LatA concentration by half (2.4 µM or 1 µg/ml) dramatically reduced actin rod frequency from at least 75% of cells to about 10%. Further reducing it (1.2 µM or 0.5 µg/ml) precluded actin rod formation altogether (see Fig. S3b).
Interestingly, most actin rods described in the literature are detectable only by antibodies against cofilin (10), while the actin rods described here were readily detectable by Lifeact and phalloidin. In our experimental conditions, an antibody against cofilin did detect a subpopulation of actin rods that were mostly needle shaped (see Fig. S4, second row, in the supplemental material). These needle-like rods accumulated Lifeact label gradually over time until the round rods disappeared and only Lifeact-labeled needle-shaped rods remained. Rows 3 and 4 in Fig. S4 show a confocal section of a cell in which Lifeact staining transforms to the needle-like phenotype. Interestingly, needle-shaped rods appeared to be less efficient in blocking capsid motility than round rods.
Actin rod formation causes intranuclear capsid motility to stop.
From the previous experiment, it was not clear if LatA-induced actin rod formation directly leads to intranuclear capsid stalling or if these phenotypes are epiphenomena. It was previously reported that CytD can inhibit actin rod formation (10) in addition to its well-established F-actin-severing activity by binding to the barbed end (23). Moreover, CytD does not have an effect on NCM (1). Therefore, we infected MEF-LA cells with capsid-tagged PRV and incubated the cells with 2 µM of CytD and 2 µg/ml of LatA for 1 h at 4 hpi. We then recorded capsid motility within the nucleus and performed ICM. We could not detect actin rod formation in these double-treated cells, and capsid motility was only slightly reduced, as measured by ICM (Fig. 4A), which might be a general cytotoxic effect.
FIG 4 .
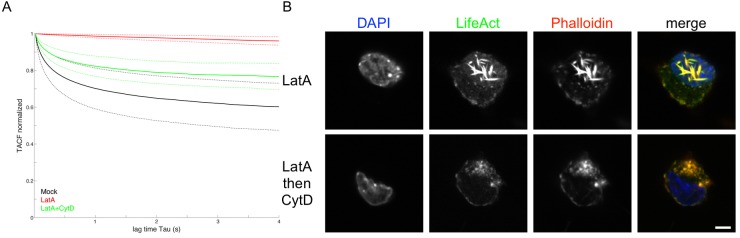
Actin rod formation causes intranuclear capsid motility to stop. MEF cells constitutively expressing Lifeact-EGFP were infected with PRV mRFP-VP26. (A) Cells at 4 hpi were either mock treated (black) or incubated for 1 h with 4.7 µM (2 µg/ml) LatA (red) or 2 µM of CytD as well as 4.7 µM (2 µg/ml) LatA (green). Oblique laser microscopy was performed with 100 frames/s for 10 s, and resulting time-lapse videos of infected nuclei were subjected to autocorrelation analysis. Solid lines indicate mean values; dashed lines indicate the means ± standard deviations (23 cells mock, 7 cells CytD and LatA). (B) Cells treated with LatA or with LatA and after 1 h with CytD were fixed, stained with DAPI and fluorescent phalloidin, and analyzed by confocal laser scanning microscopy. One confocal section is shown. Scale bar indicates 5 µm.
In addition, we incubated with LatA first, waited for the induction of actin rods, and then added CytD. Actin rods disappeared after CytD addition (Fig. 4B) in concentrations of 0.5 µM CytD or higher (see Fig. S5 in the supplemental material). We conclude that actin rod formation is the cause of LatA-induced inhibition of NCM.
Intranuclear capsids bind to LatA-induced actin rods.
As the previous result suggested a direct binding of nuclear capsids to actin rods, we next sought to show this interaction biochemically. To this end, we infected MEF-LA with capsid-tagged PRV and treated with LatA as described above. We then lysed the cells and used an anti-GFP antibody to immunoprecipitate Lifeact-GFP and associated proteins. By Western blotting, we detected Lifeact-GFP and actin in the immunoprecipitates of all conditions. However, only in the LatA-treated cells did we detect the major capsid protein VP5 (Fig. 5). Thus, we conclude that LatA-induced actin rods most likely trap intranuclear capsids by binding them, leading to the observed drop in motility by live-cell microscopy. This provides an intriguing alternative explanation of how LatA leads to intranuclear capsid stalling and provides a good model for why LatA influences capsid motility but the other tested F-actin-depolymerizing drugs do not.
FIG 5 .
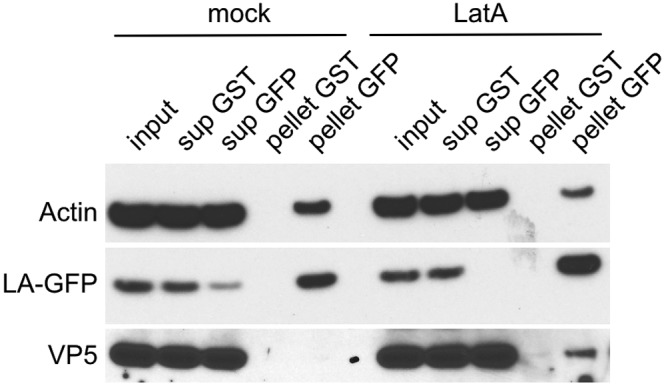
Latrunculin A-induced actin rods bind the major capsid protein. MEF cells constitutively expressing Lifeact-EGFP were infected with PRV mRFP-VP26. Cells at 4 hpi were either mock treated (mock) or incubated for 1 h with 4.7 µM (2 µg/ml) LatA (LatA). Cells were lysed, and the precleared lysates were incubated with antibodies directed against glutathione S-transferase (GST) or GFP, detecting Lifeact-EGFP (LA-GFP) and protein A/G beads for 1.5 h at 4°C. Bead-bound immunoprecipitates were separated from supernatant and washed extensively. Ten percent of lysate (input) and supernatants (sup) and 50% of immunoprecipitated protein (pellet) were subjected to SDS-PAGE and Western blotting using antiactin, anti-GFP, and anti-VP5 antibodies.
Intranuclear capsid motility is unaffected by LatA in PK15 cells, which do not form actin rods.
We tested several established cell lines for their capacity to form intranuclear actin rods. In the mouse fibroblast cell line NIH/3T3, LatA treatment blocked NCM, and we observed round actin rods in LatA-treated cells after phalloidin staining (see Fig. S6). Surprisingly, in PK15 cells, LatA treatment did not block NCM. PK15 cells are a transformed pig kidney cell line frequently used to grow PRV (Fig. 6A and B and Video S3 in the supplemental material). This observation was also true when we used up to 4-fold-higher concentrations of LatA or longer incubation times. Importantly, we could not detect intranuclear actin rods in PK15 cells treated with LatA by phalloidin (Fig. 6c), while cytoplasmic F-actin was clearly degraded by LatA. This finding is consistent with the hypothesis that LatA affects capsid motility only if actin rods are formed.
FIG 6 .

Nuclear capsid motility is unaffected by LatA in PK15 cells. PK15 cells were infected with PRV mRFP-VP26. (A and B) Cells at 4 hpi were either mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA. Intranuclear capsid motility was recorded by live-cell spinning disk microscopy with 7.4 frames/s. (A) The right column represents a temporal maximum projection of 100 frames at 7.4 frames/s. Note the degree of signal smearing, which indicates capsid motility compared to a single frame depicted in the left column. (B) Videos of infected nuclei, like the ones shown in panel A, were analyzed by ICM. Black represents mock treatment, and red represents LatA treatment. Solid lines indicate mean values; dashed lines indicate the means ± standard deviations (16 cells mock, 18 cells LatA). (C) LatA-treated and infected PK15 cells were fixed and stained with DAPI and fluorescent phalloidin. A maximum intensity projection of about 15 µm is shown. Note the absence of nuclear actin rods. Scale bars indicate 5 µm.
DISCUSSION
We provide evidence that F-actin is not involved in intranuclear capsid motility of herpesvirus capsids in primary fibroblasts. We found no indication of nuclear F-actin in cells infected with representative members of all three subfamilies of the Herpesviridae. This finding is consistent with reports showing that steady-state somatic cells do not exhibit phalloidin-stainable nuclear F-actin but that most nuclear actin is in a monomeric, G-actin form. This idea was underlined by a recent report that Lifeact- and phalloidin-stainable nuclear F-actin could be found in somatic cells only as a transient effect of serum stimulation (24). Another recent report describes submicron-length actin polymers using a truncated form of utrophin as a novel nuclear F-actin probe (25). But as the described polymers were extremely short, the authors suggested that it is unlikely they are involved in transport processes. Moreover, they also ruled out directed propulsive force generation by these novel actin polymers (7, 25).
These findings stand in contrast to those from studies with Autographa californica multiple nucleopolyhedrovirus (ACMNPV). This virus encodes a WASP (Wiskott-Aldrich syndrome protein)-like viral protein that translocates the Arp2/3 complex into the nucleus to activate actin polymerization (26). To our knowledge, no herpesvirus encodes such a WASP homolog.
Currently, our work addresses infection of primary mouse fibroblasts. We do not know what type of nuclear F-actin is formed after PRV infection of superior cervical ganglia neurons (2). These intranuclear structures cannot be labeled by Lifeact but are stained by phalloidin (unpublished results). As a result, we were unable to determine if they play a role in capsid transport. However, these structures are located primarily at the nuclear rim, while capsid motility fills most of the nucleus. Thus, it is likely that these structures do not play a role in bulk capsid motility in neurons but are a neuron-specific response to herpes infection. Also we can clearly detect nuclear capsid motility in cells that do not show any evidence of nuclear F-actin. Therefore, it is unlikely that virally induced F-actin as seen in primary neurons is the cause of the generally observed phenomenon of nuclear capsid motility.
Previous reports suggested that nuclear capsid motility was dependent on F-actin based on its susceptibility to LatA (1). In this report, we evaluated a wider selection of F-actin inhibitors for their effect on nuclear capsid motility by using image correlation analysis to extract bulk motility information from thousands of capsids. Only LatA had an effect on capsid motility, while all other drugs tested did not have a significant effect. As reported before, LatA treatment induced actin rods (22), and surprisingly these rods seemed to bind capsids, inhibiting their motility. We detected two major forms of rod structures by Lifeact/phalloidin and anticofilin immunostaining: one was round, while the other was more needle shaped. Capsid motility seemed to be more efficiently inhibited by the round form, perhaps due to greater surface area. The second needle-like form was stained by anticofilin antibodies, while the first was not. This needle-like form seems to be the form of actin rods previously described in the literature (10) as consisting of cofilin-actin complexes that form higher-order filamentous structures. The round form has not been described before and seems to be even more prevalent in NIH/3T3 cells treated with LatA. Interestingly, anticofilin antiserum detected the needle-like form early after LatA induction, while Lifeact stained these needles only later, after the round form disappeared in MEF cells. This finding is consistent with our observation by confocal microscopy that needles stained by Lifeact often showed an internal cofilin stain and an external Lifeact label, possibly indicating that Lifeact bound to the outside of these structures only after they were formed. This observation is in contrast to an earlier report that Lifeact cannot stain nuclear actin rods induced by heat shock in mouse striatal neuron-derived STHdh cells (27).
We were able to inhibit and reverse the formation of nuclear actin rods by the addition of CytD, another F-actin inhibitor, as described earlier (10). Most importantly, nuclear capsid motility was not significantly inhibited if both LatA and CytD were added together, again arguing against F-actin-dependent capsid motility.
Our microscopy data suggest that actin rods bind to capsids and block their motility. We confirmed this idea by immunoprecipitation experiments and found that the major capsid protein VP5 bound to Lifeact-EGFP and actin, but only in LatA-treated cells.
Finally, we found that PRV capsids maintained motility in LatA-treated PK15 cells. In addition, we could not find nuclear actin rods in these cells after LatA treatment. These two findings support our model that LatA treatment inhibits capsid motility only if nuclear actin rods are formed. Importantly, we observed almost a complete block of capsid motility in cells that formed nuclear actin rods, especially in cells that formed the round form of actin rods. This phenotype was observable without any sophisticated quantification method, and the ICM results reflected this strong effect. In contrast, Forest et al. (1) describe a modest 2-fold reduction of capsid velocity after LatA addition under their conditions. Moreover, effects of ATP depletion on capsid motility (1) must be taken with caution, as ATP depletion can also induce actin rods (9, 10). In addition, actin rods can also be induced by overexpressing actin fusion proteins containing a nuclear localization signal (5, 28). Therefore, conclusions on the role of actin rods in intranuclear motility should be reevaluated in light of our current results (29).
The ICM method used in this study provides an estimate of capsid mobility but cannot measure the directedness of that motion. The central finding of this study is therefore that the majority of nuclear capsid motility is independent of F-actin. The mechanism of the characteristic swirling motion of intranuclear herpes capsid motility remains an open question. One approach will be to use high-speed single-particle tracking. As nuclear capsid motility is fast and three-dimensional, microscopy with a fixed focal plane invariably results in tracks too short to provide a reliable basis for particle motility analysis. Therefore, frame rates must be fast (at least 10 frames per second or more), and methods yielding three-dimensional localization are necessary for high-fidelity single-particle tracking analysis. Moreover, it is now clear that fusions of certain fluorescent proteins, like EGFP, to the small capsid protein result in nuclear aggregation (4). However, small capsid protein fusions that show fewer aggregates, like the mRFP and mCherry fusions used here, produce many more intranuclear capsids. These fusions make single-particle tracking even more difficult and increase the need for extremely fast image acquisition. Nevertheless, with recent advances in imaging technology, it is now possible to determine if nuclear capsid motility is indeed directed and energy dependent. The time has come to reopen the question: How do nuclear capsids move?
MATERIALS AND METHODS
Cells and viruses.
MEF cells as well as PK15 cells (ATCC CCL-33) were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum under standard conditions. NIH/3T3 cells (ATCC CRL-1658) were cultured in DMEM containing 10% bovine calf serum.
PRV strain 180 expressing an mRFP-VP26 fusion is described in del Rio et al. (14). It encodes a fusion protein between mRFP and the amino terminus of VP26. HSV-1 carrying mRFP-VP26 (13) was a kind gift of Beate Sodeik (MHH Hannover, Germany). MCMV S-mCherry-SCP is described in reference 15. MHV68 mCherry-ORF65 was made analogously to MCMV S-mCherry-SCP (Bosse and Adler, unpublished).
Drugs.
Latrunculin A (Invitrogen; catalog no. L12370), cytochalasin D (Sigma-Aldrich; C2618), mycalolide B (Enzo; BML-T123-0020), ML-7 (EMD Millipore; 475880), and Jasplakinolide (Sigma-Aldrich; J4580) were diluted in cell culture-grade DMSO such that final working solutions never reached a final concentration that would induce actin rods. Cells were incubated with the drugs for 1 h in full medium and imaged or fixed directly afterward.
Immunoprecipitation.
For immunoprecipitations, lysates were obtained from uninfected or from PRV180-infected Lifeact MEF cells 5 hpi either mock treated or treated with 2 µg/ml latrunculin A for 1 h. Cell pellets were resuspended in buffer A (20 mM PIPES, 140 mM NaCl, 1 mM EGTA, 1 mM dithiothreitol [DTT], 8% sucrose, pH 6.8) supplemented with protease inhibitor (Roche) and Benzonase (Sigma) and passaged through a 27-gauge needle. Complete cell membrane and nuclear envelope disruption was confirmed by phase contrast microscopy. Soluble protein was separated from cellular debris by centrifugation, and the resulting lysate was precleared with protein A/G beads (Pierce) for 1 h at 4°C. Lysates were then incubated with new beads and polyclonal anti-glutathione S-transferase (GST) (Sigma; G1417) or anti-GFP (Invitrogen; A11122) antibody for 1.5 h at 4°C. After centrifugation and extensive washing, supernatants and pellets were subjected to standard SDS-PAGE and Western blotting using polyclonal anti-GFP (Invitrogen; A11122) and monoclonal anti-VP5 (27) and anti-beta-actin (Sigma; A1978) antibodies.
Phalloidin staining and immunofluorescence.
Cells were grown on fibronectin-coated (10 µg/ml in Dulbecco’s phosphate-buffered saline [DPBS], 1 h at 37°C) μ-Slides (Ibidi). Cells were fixed with 4% paraformaldehyde-DPBS for 10 min at 37°C and permeabilized with 0.1% Triton-DPBS for 20 min at room temperature (RT). DAPI (4′,6-diamidino-2-phenylindole) was used at a final concentration of 7 µM and Alexa Fluor-labeled phalloidin at 33 nM in 3% bovine serum albumin (BSA)-DPBS for 20 min. Stained samples cells were stored at 4°C in the dark until analysis.
Staining of actin rods with an ADF/cofilin antibody (Cytoskeleton Inc.; ACFL01) was done according to a protocol kindly communicated by James Bamburg (Colorado State University, USA), as ADF/cofilin is poor to nonexistent after 0.1% Triton permeabilization: cells were fixed for 45 min with 4% of paraformaldehyde-DPBS and permeabilized with −20°C cold methanol for 3 min. After being blocked for 1 h with 3% BSA-DPBS, the ADF/cofilin antibody was added in 3% BSA-DPBS and incubated at room temperature for 1 h. Alexa Flour-labeled secondary antibodies (Invitrogen) were used 1:1,000 in 3% BSA-DPBS and incubated at room temperature for 1 h.
Confocal laser scanning and spinning disk microscopy.
Fixed samples were imaged on a Nikon A1-RS confocal laser scanning microscope using a ×60 magnification, 1.4-numerical-aperture (NA) oil objective, and 405-, 488-, 561-, and 647-nm laser excitation. General live-cell microscopy was performed on a spinning disk microscope equipped with a ×60 magnification, 1.4-NA oil objective, and 488-, 561-, and 647-nm laser excitation. Images were collected on an electron-multiplying charge-coupled device (EMCCD) camera. Physiological conditions were kept constant at 5% CO2 and 37°C throughout the experiments.
Image acquisition for correlation microscopy.
Live imaging for image correlation microscopy was performed in oblique illumination mode using a 1.49-NA total internal-reflection fluorescence (TIRF) oil objective, except for Fig. 6, where spinning disk microscopy was used. Images were acquired on an EMCCD camera. Every imaging sequence consisted of 1,000 frames acquired at 100 frames/s for 10 s for oblique microscopy or 100 frames acquired at 7.4 frames/s for Fig. 6. Cells were grown on fibronectin-coated culture dishes. Dishes had 7-mm openings and 25-mm round coverslips glued onto them as a custom option (MatTek) to provide extra rigidity. A heated microscopy stage and an objective heater were used to maintain physiological conditions. Images were cropped (approximately 64 by 64 pixels) so that they represented only one nucleus per data set.
Image manipulation.
Image manipulation was done in ImageJ/Fiji in accordance with mBio guidelines (30). Only linear enhancements were performed. Figures were arranged in Adobe Illustrator and Pixelmator.
Image autocorrelation.
Quantification of capsid motility by ICM was done according to Gaborski et al. (19) with a custom written MatLab (MathWorks) script (see Text S1 in the supplemental material for a sample code). Briefly, the degree of motility of fluorescent features in a time-lapse image sequence can be expressed by using the temporal autocorrelation function (TACF). Images of slow-moving particles taken after brief periods differ only slightly and therefore still show a high level of correlation. Correlation over long lag times decreases as particles move away from each other. Rapidly moving particles result in faster decay of the temporal autocorrelation function.
SUPPLEMENTAL MATERIAL
MEF cells constitutively expressing Lifeact-EGFP were infected with HSV-1 mRFP-VP26, PRV mRFP-VP26, MCMV S-mCherry-SCP, or MHV-68 mCherry-ORF65 at an MOI of at least 5. Live-cell spinning disk microscopy with at least 3.7 frames/s was performed in one confocal section after the first capsids appeared in the nucleus at 6 hpi, 4 hpi, 20 hpi, or 8 hpi. Videos in this montage represent HSV-1-, PRV-, MCMV-, and MHV-68-infected cells from left to right. Each video represents a 5.4-s acquisition time. Scale bar indicates 5 µm. Download
MEF cells constitutively expressing Lifeact-EGFP were infected with PRV mRFP-VP26 at an MOI of 10. Cells at 4 hpi were either mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA and imaged by live-cell spinning disk microscopy in one confocal section. Fifty frames acquired at 3.7 frames/s are shown. Scale bar indicates 5 µm. Download
PK15 cells were infected with PRV mRFP-VP26 at an MOI of 10. Cells at 4 hpi, infected with PRV mRFP-VP26, were either mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA and imaged by live-cell spinning disk microscopy in one confocal section. One-hundred frames acquired at 7.4 frames/s are shown. Scale bar indicates 5 µm. Download
Effect of various concentrations of jasplakinolide and mycalolide B on F-actin. MEF cells constitutively expressing Lifeact-EGFP (white) were treated with various concentrations of jasplakinolide and mycalolide B for 1 h, fixed, counterstained with DAPI (blue), and imaged by confocal laser scanning microscopy in one confocal section. Scale bars indicate 10 µm. Download
Effect of various concentrations of DMSO on F-actin. MEF cells constitutively expressing Lifeact-EGFP (white) were treated with various concentrations of DMSO for 1 h, fixed, counterstained with DAPI (blue), and imaged by confocal laser scanning microscopy in one confocal section. Scale bar indicates 10 µm. Download
Effect of various concentrations of DMSO on F-actin. (a) MEF cells (MEF) or MEF cells constitutively expressing Lifeact-EGFP (MEF-Lifeact) were treated with various concentrations of latrunculin A for 1 h, fixed, and stained with fluorescently labeled phalloidin as well as with DAPI (blue) and imaged by confocal laser scanning microscopy in one confocal section. In pictures showing merged channels, Lifeact is coded green and phalloidin is coded red, while DAPI is coded in blue. (b) MEF cells as well as MEF cells expressing Lifeact were treated with various concentrations of latrunculin A as described above, and the frequency of nuclear actin rod appearance was counted. LatA at 2 µg/ml corresponds to 4.7 µM. Download
Two different kinds of actin rods are induced by LatA. MEF cells constitutively expressing Lifeact-EGFP were mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA, fixed with paraformaldehyde, and then extracted with ice-cold methanol. Afterward, cells were stained with a polyclonal antiserum directed against cofilin and counterstained with DAPI. Images were acquired by laser scanning confocal microscopy in one confocal section. The scale bar indicates 5 µm. Download
Cytochalasin D can revert actin rod formation by LatA. MEF cells constitutively expressing Lifeact-EGFP (white) were treated with 4.7 µM (2 µg/ml) latrunculin A for 1 h. Then, various concentrations of cytochalasin D were added for 1 h and cells were fixed, counterstained with DAPI (blue), and imaged by confocal laser scanning microscopy in one confocal section. The scale bar indicates 10 µm. Download
LatA induces actin rods NIH/3T3 cells. NIH/3T3 cells were mock infected or infected with PRV mRFP-VP26. Cells at 4 hpi were incubated for 1 h with 4.7 µM (2 µg/ml) LatA. Cells were then fixed and stained with fluorescently labeled phalloidin as well as with DAPI and imaged by confocal laser scanning microscopy in one confocal section. Scale bars indicate 10 µm. Download
MatLab script used for autocorrelation analysis. Download
ACKNOWLEDGMENTS
We thank Michael Sixt (Institute of Science and Technology, Klosterneuburg, Austria) for the generous gift of Lifeact-expressing mice, Torsten Sacher (Max von Pettenkofer Institute, Munich, Germany) for help with MEF preparation, James Bamburg (Colorado State University, Fort Collins, CO) for help with anticofilin stains, and Beate Sodeik (MHH, Hannover, Germany) for HSV-1-expressing mRFP-VP26. We also thank Halina Staniszewska Goraczniak for excellent technical support and Gary S. Laevsky (Princeton Molecular Biology Imaging Facility) for imaging support.
This work was supported by National Institutes of Health (NIH) grants NS033506 and NS060699. The Imaging Core Facility at the Lewis-Sigler Institute is funded by NIH National Institute of General Medical Sciences Center grant PM50 GM071508. J.B.B. was supported by a postdoctoral research fellowship from the German Research Foundation (DFG).
Footnotes
Citation Bosse JB, Virding S, Thiberge SY, Scherer J, Wodrich H, Ruzsics Z, Koszinowski UH, Enquist LW. 2014. Nuclear herpesvirus capsid motility is not dependent on F-actin. mBio 5(5):e01909-14. doi:10.1128/mBio.01909-14.
REFERENCES
- 1. Forest T, Barnard S, Baines JD. 2005. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 7:429–431. 10.1038/ncb1243 [DOI] [PubMed] [Google Scholar]
- 2. Feierbach B, Piccinotti S, Bisher M, Denk W, Enquist LW. 2006. Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog. 2:e85. 10.1371/journal.ppat.0020085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ward PL, Ogle WO, Roizman B. 1996. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70:4623–4631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagel C-H, Döhner K, Binz A, Bauerfeind R, Sodeik B. 2012. Improper tagging of the non-essential small capsid protein VP26 impairs nuclear capsid egress of herpes simplex virus. PLoS One 7:e44177. 10.1371/journal.pone.0044177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Lanerolle P, Serebryannyy L. 2011. Nuclear actin and myosins: life without filaments. Nat. Cell Biol. 13:1282–1288. 10.1038/ncb2364 [DOI] [PubMed] [Google Scholar]
- 6. Grosse R, Vartiainen MK. 2013. To be or not to be assembled: progressing into nuclear actin filaments. Nat. Rev. Mol. Cell Biol. 14:693–697. 10.1038/nrm3681 [DOI] [PubMed] [Google Scholar]
- 7. Belin BJ, Mullins RD. 2013. What we talk about when we talk about nuclear actin. Nucleus 4:291–297. 10.4161/nucl.25960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukui Y, Katsumaru H. 1979. Nuclear actin bundles in amoeba, dictyostelium and human HeLa cells induced by dimethyl sulfoxide. Exp. Cell Res. 120:451–455. 10.1016/0014-4827(79)90412-9 [DOI] [PubMed] [Google Scholar]
- 9. Bernstein BW, Chen H, Boyle JA, Bamburg JR. 2006. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am. J. Physiol. Cell Physiol. 291:C828–C839. 10.1152/ajpcell.00066.2006 [DOI] [PubMed] [Google Scholar]
- 10. Minamide LS, Maiti S, Boyle JA, Davis RC, Coppinger JA, Bao Y, Huang TY, Yates J, Bokoch GM, Bamburg JR. 2010. Isolation and characterization of cytoplasmic cofilin-actin rods. J. Biol. Chem. 285:5450–5460. 10.1074/jbc.M109.063768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang L, Godinez WJ, Kim I-H, Tektonidis M, de Lanerolle P, Eils R, Rohr K, Knipe DM. 2011. PNAS plus: herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc. Natl. Acad. Sci. U. S. A. 108:E136–E144. 10.1073/pnas.1103411108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehta IS, Amira M, Harvey AJ, Bridger JM. 2010. Rapid chromosome territory relocation by nuclear motor activity in response to serum removal in primary human fibroblasts. Genome Biol. 11:R5. 10.1186/gb-2010-11-s1-p5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagel CH, Döhner K, Fathollahy M, Strive T, Borst EM, Messerle M, Sodeik B. 2008. Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J. Virol. 82:3109–3124. 10.1128/JVI.02124-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. del Rio T, Ch’ng TH, Flood EA, Gross SP, Enquist LW. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 79:3903–3919. 10.1128/JVI.79.7.3903-3919.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bosse JB, Bauerfeind R, Popilka L, Marcinowski L, Taeglich M, Jung C, Striebinger H, von Einem J, Gaul U, Walther P, Koszinowski UH, Ruzsics Z. 2012. A beta-herpesvirus with fluorescent capsids to study transport in living cells. PLoS One 7:e40585. 10.1371/journal.pone.0040585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5:605–607. 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riedl J, Flynn KC, Raducanu A, Gärtner F, Beck G, Bösl M, Bradke F, Massberg S, Aszodi A, Sixt M, Wedlich-Söldner R. 2010. Lifeact mice for studying F-actin dynamics. Nat. Methods 7:168–169. 10.1038/nmeth0310-168 [DOI] [PubMed] [Google Scholar]
- 18. Coué M, Brenner SL, Spector I, Korn ED. 1987. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213:316–318. 10.1016/0014-5793(87)81513-2 [DOI] [PubMed] [Google Scholar]
- 19. Gaborski TR, Sealander MN, Ehrenberg M, Waugh RE, McGrath JL. 2010. Image correlation microscopy for uniform illumination. J. Microsc. 237:39–50. 10.1111/j.1365-2818.2009.03300.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saito S, Watabe S, Ozaki H, Fusetani N, Karaki H. 1994. Mycalolide B, a novel actin depolymerizing agent. J. Biol. Chem. 269:29710–29714 [PubMed] [Google Scholar]
- 21. Bubb MR, Spector I, Beyer BB, Fosen KM. 2000. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 275:5163–5170. 10.1074/jbc.275.7.5163 [DOI] [PubMed] [Google Scholar]
- 22. Pendleton A, Pope B, Weeds A, Koffer A. 2003. Latrunculin B or ATP depletion induces cofilin-dependent translocation of actin into nuclei of mast cells. J. Biol. Chem. 278:14394–14400. 10.1074/jbc.M206393200 [DOI] [PubMed] [Google Scholar]
- 23. Brenner SL, Korn ED. 1980. The effects of cytochalasins on actin polymerization and actin ATPase provide insights into the mechanism of polymerization. J. Biol. Chem. 255:841–844 [PubMed] [Google Scholar]
- 24. Baarlink C, Wang H, Grosse R. 2013. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 340:864–867. 10.1126/science.1235038 [DOI] [PubMed] [Google Scholar]
- 25. Belin BJ, Cimini BA, Blackburn EH, Mullins RD. 2013. Visualization of actin filaments and monomers in somatic cell nuclei. Mol. Biol. Cell 24:982–994. 10.1091/mbc.E12-09-0685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goley ED, Ohkawa T, Mancuso J, Woodruff JB, D’Alessio JA, Cande WZ, Volkman LE, Welch MD. 2006. Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science 314:464–467. 10.1126/science.1133348 [DOI] [PubMed] [Google Scholar]
- 27. Munsie LN, Caron N, Desmond CR, Truant R. 2009. Lifeact cannot visualize some forms of stress-induced twisted F-actin. Nat. Methods 6:317. 10.1038/nmeth0509-317 [DOI] [PubMed] [Google Scholar]
- 28. Kokai E, Beck H, Weissbach J, Arnold F, Sinske D, Sebert U, Gaiselmann G, Schmidt V, Walther P, Münch J, Posern G, Knöll B. 2014. Analysis of nuclear actin by overexpression of wild-type and actin mutant proteins. Histochem. Cell Biol. 141:123–135. 10.1007/s00418-013-1151-4 [DOI] [PubMed] [Google Scholar]
- 29. Chang L, Godinez WJ, Kim IH, Tektonidis M, de Lanerolle P, Eils R, Rohr K, Knipe DM. 2011. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc. Natl. Acad. Sci. USA 108:E136–E144. 10.1073/pnas.1103411108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MEF cells constitutively expressing Lifeact-EGFP were infected with HSV-1 mRFP-VP26, PRV mRFP-VP26, MCMV S-mCherry-SCP, or MHV-68 mCherry-ORF65 at an MOI of at least 5. Live-cell spinning disk microscopy with at least 3.7 frames/s was performed in one confocal section after the first capsids appeared in the nucleus at 6 hpi, 4 hpi, 20 hpi, or 8 hpi. Videos in this montage represent HSV-1-, PRV-, MCMV-, and MHV-68-infected cells from left to right. Each video represents a 5.4-s acquisition time. Scale bar indicates 5 µm. Download
MEF cells constitutively expressing Lifeact-EGFP were infected with PRV mRFP-VP26 at an MOI of 10. Cells at 4 hpi were either mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA and imaged by live-cell spinning disk microscopy in one confocal section. Fifty frames acquired at 3.7 frames/s are shown. Scale bar indicates 5 µm. Download
PK15 cells were infected with PRV mRFP-VP26 at an MOI of 10. Cells at 4 hpi, infected with PRV mRFP-VP26, were either mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA and imaged by live-cell spinning disk microscopy in one confocal section. One-hundred frames acquired at 7.4 frames/s are shown. Scale bar indicates 5 µm. Download
Effect of various concentrations of jasplakinolide and mycalolide B on F-actin. MEF cells constitutively expressing Lifeact-EGFP (white) were treated with various concentrations of jasplakinolide and mycalolide B for 1 h, fixed, counterstained with DAPI (blue), and imaged by confocal laser scanning microscopy in one confocal section. Scale bars indicate 10 µm. Download
Effect of various concentrations of DMSO on F-actin. MEF cells constitutively expressing Lifeact-EGFP (white) were treated with various concentrations of DMSO for 1 h, fixed, counterstained with DAPI (blue), and imaged by confocal laser scanning microscopy in one confocal section. Scale bar indicates 10 µm. Download
Effect of various concentrations of DMSO on F-actin. (a) MEF cells (MEF) or MEF cells constitutively expressing Lifeact-EGFP (MEF-Lifeact) were treated with various concentrations of latrunculin A for 1 h, fixed, and stained with fluorescently labeled phalloidin as well as with DAPI (blue) and imaged by confocal laser scanning microscopy in one confocal section. In pictures showing merged channels, Lifeact is coded green and phalloidin is coded red, while DAPI is coded in blue. (b) MEF cells as well as MEF cells expressing Lifeact were treated with various concentrations of latrunculin A as described above, and the frequency of nuclear actin rod appearance was counted. LatA at 2 µg/ml corresponds to 4.7 µM. Download
Two different kinds of actin rods are induced by LatA. MEF cells constitutively expressing Lifeact-EGFP were mock treated or incubated for 1 h with 4.7 µM (2 µg/ml) LatA, fixed with paraformaldehyde, and then extracted with ice-cold methanol. Afterward, cells were stained with a polyclonal antiserum directed against cofilin and counterstained with DAPI. Images were acquired by laser scanning confocal microscopy in one confocal section. The scale bar indicates 5 µm. Download
Cytochalasin D can revert actin rod formation by LatA. MEF cells constitutively expressing Lifeact-EGFP (white) were treated with 4.7 µM (2 µg/ml) latrunculin A for 1 h. Then, various concentrations of cytochalasin D were added for 1 h and cells were fixed, counterstained with DAPI (blue), and imaged by confocal laser scanning microscopy in one confocal section. The scale bar indicates 10 µm. Download
LatA induces actin rods NIH/3T3 cells. NIH/3T3 cells were mock infected or infected with PRV mRFP-VP26. Cells at 4 hpi were incubated for 1 h with 4.7 µM (2 µg/ml) LatA. Cells were then fixed and stained with fluorescently labeled phalloidin as well as with DAPI and imaged by confocal laser scanning microscopy in one confocal section. Scale bars indicate 10 µm. Download
MatLab script used for autocorrelation analysis. Download