

Abstract
Growing evidence suggests that tumor-associated macrophages (TAMs) promote cancer progression and therapeutic resistance by enhancing angiogenesis, matrix-remodeling and immunosuppression. In this study prostate cancer (PCa) under androgen blockade therapy (ABT) was investigated, demonstrating that TAMs contribute to PCa disease recurrence through paracrine signaling processes. ABT induced the tumor cells to express macrophage colony-stimulating factor 1 (M-CSF-1 or CSF-1) and other cytokines that recruit and modulate macrophages, causing a significant increase in TAM infiltration. Inhibitors of CSF-1 signaling through its receptor, CSF-1R, were tested in combination with ABT, demonstrating that blockade of TAM influx in this setting disrupts tumor promotion and sustains a more durable therapeutic response compared to ABT alone.
Introduction
Androgen blockade therapy (ABT) for treating prostate cancer (PCa) was initially conceived through the discovery by Dr. Huggins and colleagues that PCa growth is dependent on androgens (1), and this now has become a standard treatment. Over the years, pharmacological interventions that disrupt either androgen biosynthesis or the androgen receptor (AR) have been developed to treat PCa. Two new drugs approved by the FDA in 2012, Abiraterone (Zytiga) and MDV3100 (Enzalutamide or Xtandi) that effectively block either the androgen synthesis enzyme, CYP17, or AR ligand binding, respectively, have energized the ABT field (2,3). Both agents prolong the overall survival of patients with castration-resistant prostate cancer (CRPC). However PCa treated with these new agents also can acquire resistance through amplified AR expression, aberrant activation of AR by tyrosine kinase signaling, atypical activation of AR co-activators, and AR splice variants (3–7), thus sustaining the need for improved treatments for this indication.
A less studied, but likely important, aspect of therapeutic resistance is the influence of the tumor microenvironment on ABT resistance (8). Tumor-associated macrophages (TAMs) often constitute a significant inflammatory component in the tumor, and have been shown to promote tumor progression and resistance to various chemotherapeutic agents (9,10). The recruitment and functional evolution of macrophages from systemic sites to the tumor environment is a complex process that is dictated by various cytokines, tissue factors, and conditions (11). TAMs have been described to exist in different activation states, ranging from classically activated M1 macrophages, which are proposed to be anti-tumorigenic, to alternatively activated M2 macrophages, which are reported to be pro-tumorigenic (11). Proposed mechanisms by which M2-TAMs can promote tumor progression include suppressing the adaptive immune response against cancer cells, promoting tumor growth through angiogenesis, or secreting tumorigenic growth factors (12,13). A prominent cytokine known to regulate myeloid development, macrophage differentiation, and proliferation is the macrophage colony stimulating factor (M-CSF or CSF-1) (14). CSF-1-mediated signaling has been shown to be critical for the recruitment of TAMs to tumors, and also to skew them towards the M2 phenotype (14–16).
The role of TAMs in PCa progression, and more specifically in the context of ABT, is not well understood. A recent clinical study showed that the infiltration of CD68+ macrophages was increased in tumor biopsy samples taken from patients who had received ABT and this increase in TAMs is correlated with time to tumor progression (17). In a preclinical study, surgical castration of mice bearing murine Myc-CaP tumors resulted in increased influx of inflammatory cells, including B cells, natural killer (NK) cells, and macrophages (18). This study emphasized B cells as key contributors to the emergence of CRPC, but their data showed that TAMs are the major immune cells in the tumor and they also increased after castration (18). To gain a better understanding of the protumorigenic role of TAMs in the context of anti-androgen therapy, we used the androgen-dependent and immunocompetent Myc-CaP tumor and intraprostatic CWR22Rv1 xenograft model, as the primary and secondary model respectively to investigate this issue. We found that ABT, either by castration or MDV3100 treatment, induced cytokine expression in tumor cells, which, in turn, promoted a pro-tumorigenic M2 phenotype in TAMs. These findings suggest that the incorporation of a TAM inhibition regimen, such as CSF1R blockade, could improve the efficacy and durability of ABT for PCa.
Materials and Methods
Cell Culture and drugs
The murine macrophage RAW264.7 (RAW) cells (ATCC), and Myc-CaP cells (a kind gift from Dr. Charles Sawyers, Memorial Sloan Kettering New York) were cultured in DMEM, while LNCAP, LNCaP-C4-2 (ATCC), and CWR22Rv1 (kind gift from Dr. David Agus, Cedars-Sinai Medical Center) cells were cultured in RPMI medium. Both media were supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (P/S). The charcoal stripped serum (CSS) used was charcoal dextran treated FBS (Omega Scientific Inc.). GW2580 (LC Labs) was diluted in DMSO. PLX3397, 5-[(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)methyl]-N-[[6-(trifluoromethyl)-3-pyridyl]methyl]pyridin-2-amine (see Supplemental Figure 6), was synthesized at Plexxikon Inc. The detailed synthetic procedure is presented elsewhere (Tap et al. NEJM; in review).
In vitro Migration and Co-Culture Assay
RAW macrophages (1.0 × 105 cells) were seeded in 8 μm transwell inserts (BD Falcon), and placed in 24-well plates with conditioned media from Myc-CaP cells treated with 10 μM MDV3100 or DMSO vehicle. The number of migrated cells was scored after 6 hrs incubation at 37°C by 3% paraformaldehyde (PFA) fixation and stained with 4,6-diamidino-2- phenylindole (DAPI). At least 10 fields/well at 4× magnification were quantified using ImageJ Version 1.34s (NIH). To block CSF-1-signaling, we added GW2580 (1000 nM) to the top chamber containing the RAW cells.
For co-culture studies, RAW (1.0 × 106 cells) were seeded in 4 μm transwell inserts and placed in 6 well plates containing Myc-CaP (2.5 × 105 cells) treated with MDV (10 μM) or vehicle (DMSO). Total cellular RNA was extracted from co-cultured cells after 48 hrs.
Real-time RT-PCR and ELISA
Tumor cells were lysed in RIPA buffer (Upstate) containing proteinase inhibitor cocktail (Sigma), and centrifuged 5 min at 1,500 × g. Total cellular RNA was extracted according to TRIzol protocol. Real-time quantitative reverse-transcribed polymerase chain reaction (RT-PCR) was performed as previously described (19). CSF-1 levels in cell lysates and serum samples were measured using enzyme-linked immunosorbent assay (ELISA) according to the mouse CSF-1 ELISA Duoset Kit (R &D systems) with capture antibody (MAB416, R&D Systems, 2 μg/ml) and detection antibody (BAF416, R&D Systems, 0.2 μg/ml).
Tumor models
All animal experiments were approved by the Animal Research Committee of the University of California, Los Angeles. For Myc-CaP tumors, FVB male mice (6–8 weeks old, Taconic Farms) were implanted subcutaneously with 2 × 106 Myc-CaP cells. Mice were castrated when tumors reached 300–500 mm3. For PLX3397 studies, mice were fed daily chow containing PLX3397 or control chow formulated to provide an average dose of 40 mg/kg/day. Tumor size was measured every 2–3 days by digital calipers as previously described (18). Mice were sacrificed and tissues were analyzed at the ethical tumor size limit of 1 cm in diameter.
For CWR22Rv1 orthotopic tumor model, male scid-beige mice (6–8 weeks old,Taconic Farms) were implanted with 5×104 firefly luciferase marked CWR22Rv1 cells in PBS mixed 1:1 with growth factor reduced matrigel (10ul total volume), as previously described (20). Following 2 weeks of tumor establishment, the growth of tumor was assessed by bioluminescent imaging on an IVIS cooled CCD camera as previously described (20). PLX3397 treatments in this model were administered by oral gavage at a dose of 50mg/kg daily.
Immunohistochemistry
Tumor sections were harvested and fixed in 3% PFA overnight, then placed in 50% ethanol (EtOH) until paraffin embedding. Tumor sections (4 μm) were stained with F4/80 (1:500; Serotec), MMP-9 (1:1000; Abcam), CSF1R (1:200; Santa Cruz) CSF1R-Y723 (1:50; Santa Cruz), Ki67 (1:500, Vector Labs). Histological images were taken by the Nikon Eclipse 90i microscope. Five to six fields per slide of the Ki67 samples were analyzed at 10× magnification and quantified using ImageJ Version 1.34s (National Institute of Health). IHC and IHF slides were scanned by the Aperio and Arial whole slide scanner, respectively, as service provided by the UCLA Translational Pathology Core Laboratory (TPCL). Quantification of staining was analyzed by the Definiens image analysis software.
Prostate Cancer Patients and Tissue Microarray (TMA) Analyses
Prostate cancer tissues were retrieved from patients who underwent radical prostatectomy (RP) at Laval University between 1990 and 1999 and who received neoadjuvant ABT. All patients included in this study gave informed consent for tissue use and for molecular analysis. A total of 66 patients were available for the study. Patients were treated with LHRH-agonists and/or antiandrogen for a median duration of 92 days before RP. For each case included in the analysis, 6 representative prostate cancer cores (3 from primary Gleason pattern and 3 from secondary Gleason pattern) were used for TMA construction. The hormone-naïve (no treatment) TMA is a subset of over three hundred cases of prostatectomy specimens that contained representative cancer and benign areas for each case. The construction of the TMA and its use has been described previously (21,22). One 5-μm section from TMA blocks was used for IHC.
TMA sections were deparaffinized, treated with heat-induced epitope retrieval (HIER) and incubated either overnight with mouse anti-human CD68 primary antibody (1:200; Dako) or 45 minutes at room temperature with mouse monoclonal CD163 primary antibody (1:100; Cell Marque, Clone MRQ-26, #163M-16). Anti-mouse secondary antibody (Dakocytomation Envision System Labelled Polymer HRP anti-mouse, cat #K4001) was applied for 30 minutes at room temperature. Diaminobenzidine was then applied for 10 minutes at and counterstained with hematoxylin, dehydrated, and coverslipped.
Flow Cytometry
Single cell suspensions from harvested tissues were prepared for flow cytometry as previously described (23). After red blood cell (RBC) lysis (Sigma), single-cell suspensions were incubated for 30 minutes on ice with the following antibodies: CD45-APC, CD11b-APC or CD11b-e450, Gr-1-PerCPCy5.5, Ly6C-FITC, F4/80-PE-Cy7 or F4/80-e450, MHCII-Alexa-700, and CD115 (CSF1R)-PE conjugated antibodies, 1:200 (eBioscience), followed by two washes with 2% FBS in PBS (FACS buffer). Cells were fixed in 3% PFA for 15 min at room temperature and washed two times with FACS buffer. Cell acquisition was done on a BD LSR-II flow cytometer (Beckman Coulter). Data were analyzed with FlowJo software (TreeStar) (23).
Statistical Analysis
Data are presented as mean plus or minus SEM. Statistical comparisons between groups were performed using the Student t test.
Results
Androgen blockade therapy induces tumor-associated macrophage infiltration in PCa patients and a murine model of PCa
To further affirm the recent reports that ABT could be promoting TAM infiltration, we examined this issue in human PCa tissue microarrays (TMA), generated from radical prostatectomy specimens procured from PCa patients who were either treatment naïve or had received neoadjuvant hormonal ablation treatment prior to their surgery. The content of TAMs in the tissue was assessed by immunohistochemical stains against the M2 macrophage-selective marker CD163 (hemoglobin/haptoglobin scavenger receptor) (Figure 1a) and pan macrophage marker CD68 (data not shown). Tumor tissues from hormone ablation treated patients displayed a significantly higher level of CD163+ macrophages than the hormone naïve tissues (Figure 1b).
Figure 1. TAMs are elevated by ABT in PCa.
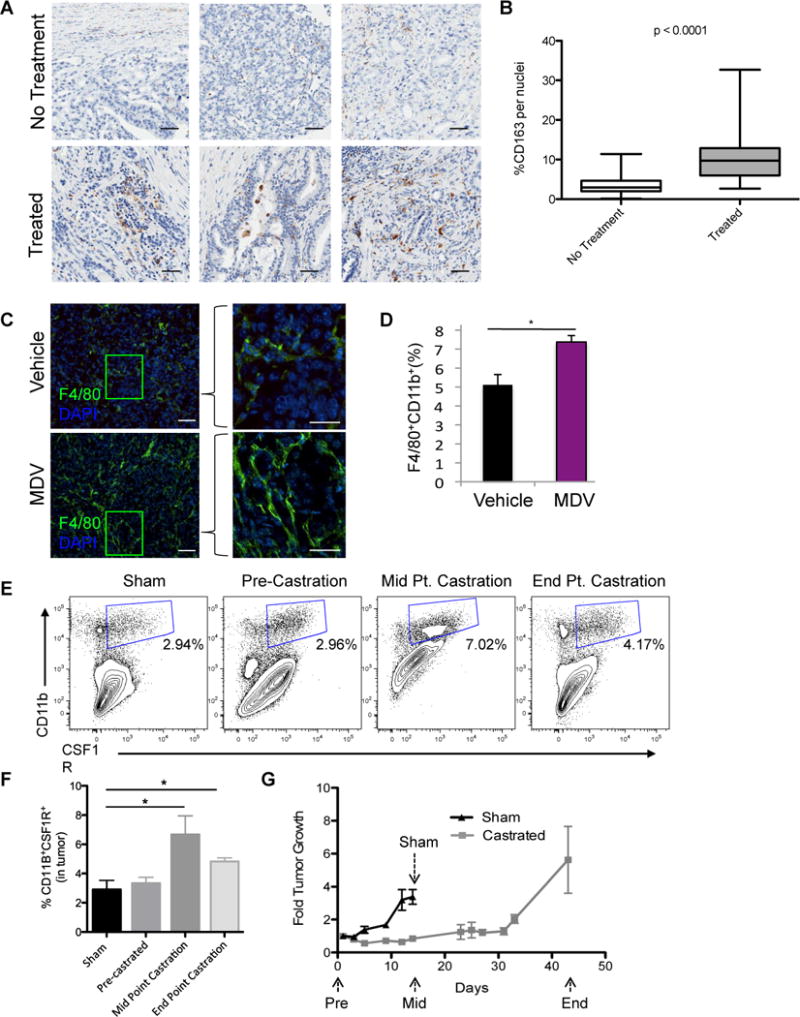
Tissue microarrays (TMAs), generated from radical prostatectomy specimens from patients who either did not (No Treatment) or did receive androgen deprivation therapy (Treated) prior to their surgery, were IHC stained for CD163. A) Representative images of the CD163 staining. Scaled bars represent 100 μm. B) Boxplots of CD163-positive cells (mean ± SD: No treatment, 3.6 ± 2.5 (n = 72); Treated, 10.7 ± 6.1 (n = 66). The impact of ABT was examined in subcutaneously implanted Myc-CaP tumors. Tumor bearing mice were treated with MDV3100 (10 mg/kg daily) orally or surgical castration when tumors reached 300–500 mm3. C) Representative IHF staining of F4/80 (green) macrophages and DAPI (blue) in Myc-CaP tumors after vehicle or MDV3100 treatment for 9 days. Scaled bars represent 100 μm (left), and 200 μm (right). D) Quantification of CD11b+F4/80+ macrophages by flow cytometry in disrupted tumors (as described in C). E) The level of CD11b+CSF1R+ macrophages in Myc-CaP tumors at specified time point after castration. Representative flow cytometry plots were shown. F, G) Quantification of CD11b+CSF1R+ macrophages in (F) and the tumor growth of (G) Myc-CaP tumors after castration. Tumor growth was expressed as fold over start of treatment (day 0). *P < 0.05. (n = 3–6 per group).
Next we turned to the implantable Myc-CaP murine prostate tumor to investigate the role of TAMs in ABT. This model offers several advantages to study this issue. First, the immunocompetent environment of this model enables the examination of both the adaptive and innate arms of the immune system in PCa progression (18). Second, the androgen signaling axis, including AR splice variants, plays a prominent role in the oncogenic progression of this model (24,25). Furthermore, macrophages comprise a significant component of the tumor microenvironment in implanted Myc-CaP tumors, as well as in the parental transgenic Hi-Myc spontaneous prostate cancer model (Supplemental Figure 1a). Detailed characterization of the different myeloid cell subsets in Myc-CaP tumors revealed that F4/80+CD11b+ TAMs predominate (2–5% of viable cells in the tumor) over CD11b+Gr-1+ MDSCs, comprising only about 0.42% of viable cells (Supplemental Figure 1a–d). As expected, the F4/80+ TAMs uniformly expressed CSF1R (CD115, c-fms), with 97.7% concordant expression between these two markers (Supplemental Figure 1c–d). Since the CSF1R+ population denotes an immune suppressive and protumorigenic myeloid cell population (26), and is also the putative targeted population of the small molecule CSF1R kinase inhibitors used in this study, the TAM population will be defined in these studies as CD11b+CSF1R+.
Two forms of ABT, namely AR inhibition with MDV3100 or androgen deprivation therapy (ADT) by surgical castration, were examined in the Myc-CaP model. A significantly higher number of F4/80+ TAMs are present in the tumor after MDV3100 treatment as assessed by immunofluorescent stain (Figure 1c) or flow cytometry (Figure 1d) as well as by qRT-PCR for F4/80 transcript from whole tumor (data not shown). Longitudinal analyses during ADT by surgical castration showed a significant increase of CD11b+CSF1R+ TAMs in the castrated group of tumors (Figure 1e, f). Referencing the influx of TAMs with respect to the tumor’s treatment response to ADT (Figure 1g), the number of TAMs peaked at 14 days after castration (midpoint) at the maximal response to ADT (i.e. the nadir of tumor growth) and declined slightly at the end point (day 42) after the tumor has regrown (Figure 1f).
These clinical and preclinical data are consistent with our postulate that ABT causes tumor cell death and injury which produces signals that recruit macrophages and modulate their functions in the tumor.
ABT induces CSF-1 expression in prostate cancer cells
We used cultured Myc-CaP tumor cells to investigate the molecular signals induced by ABT that could recruit and modulate the function of TAMs. Myc-CaP tumor cells were treated with MDV3100 or grown in androgen-deprived conditions with charcoal-striped serum (CSS) containing media (Figure 2a). The expression of 4 known macrophage-recruiting cytokines (CCL-2 (MCP-1), SDF-1, IL-34 and CSF-1) in ABT settings was examined (16). Both MDV3100 and CSS treatment significantly increased expression of CSF-1, and to a lesser extent IL-34, the second known CSF1R ligand, but not CCL-2 (MCP-1) or SDF-1, both of which were expressed at very low levels (Figure 2a). The elevated CSF-1 expression was further confirmed at the protein level (Figure 2b). The ability of ABT to induce CSF-1 expression was also observed in LNCaP and LNCaP-C4-2 human prostate cancer cells (Figure 2c, d). To assess the functional significance of the elevated CSF-1 expression, we showed that the migration of RAW264.7 (RAW) macrophage cells was enhanced by conditioned media from Myc-CaP cells treated with MDV3100, compared to those treated with vehicle (Figure 2e). Furthermore, treatment with a highly selective CSF1R inhibitor, GW2580 (23), abrogated the stimulatory effect of ABT-treated tumor conditioned media on RAW cells. Next CSF-1 expression in the ABT-treated tumors was examined. As shown in Figure 2f, a significant increase in CSF-1 mRNA was observed in Myc-CaP tumors after MDV3100 treatment (Figure 2f). Surgical castration not only induced CSF-1 expression in the tumor (Figure 2g), but CSF-1 protein level in the sera of castrated mice also increased in a time-dependent manner from day 14 to day 38 post-castration (Figure 2h). These results demonstrate that CSF-1 expression is induced by ABT of prostate tumor and it could be a key cytokine responsible for the heightened recruitment of macrophages to prostate tumors after ABT.
Figure 2. ABT induces CSF-1 expression, which promotes macrophage function.
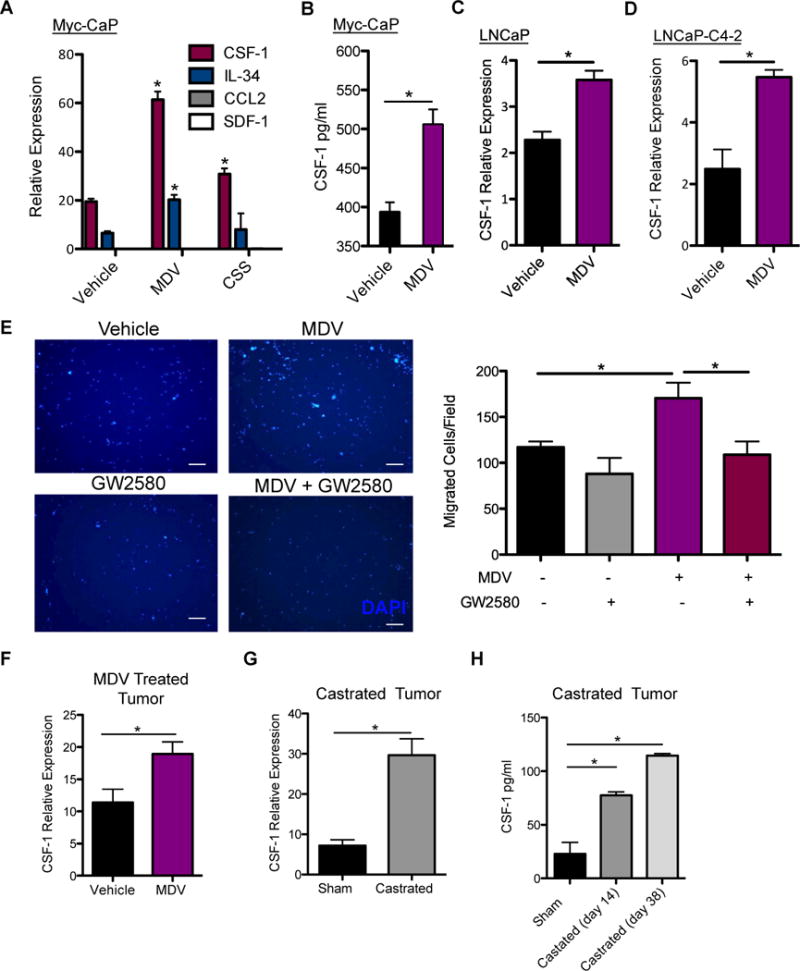
A) Quantitative RT-PCR analysis of TAM recruiting cytokines expression in Myc-CaP cells after 48 hrs of MDV3100 (10 μM) treatment. B) CSF-1 protein expression in Myc-CaP lysates after MDV3100 treatment. Quantitative RT-PCR of CSF-1 expression after 48 hrs of MDV treatment of C) LNCaP D) LNCaP-C4-2. (n = 3). E) 6 hr migration assay using RAW264.7 murine macrophages stimulated by conditioned media from Myc-CaP cells treated with MDV3100 or vehicle (DMSO), without or with the addition of 1 nM GW2580. DAPI staining of migrated RAW cells (left), migrated cell quantification of 10 fields/well at 4× magnification. Scaled bars represent 100 μm. (n = 3–6 per group). F) CSF-1 expression by RT-PCR from vehicle and MDV- treated Myc-CaP tumors. Tumor bearing mice were treated by daily oral gavage with vehicle, or MDV3100 (10 mg/kg) for 9 days. G) CSF-1 expression in Myc-CaP tumors by RT-PCR from sham (day 14 after sham surgery) and castrated (day 36 post castration) tumor bearing mice. H) Sera CSF-1 protein level analyzed by ELISA from sham and castrated mice at sham (day 14 after sham surgery), midpoint (day 14 after castration) and end point (day 38 after castration) was shown. *P < 0.05.
ABT promoted the protumorigenic phenotype of macrophages
Extensive evidence suggests that tumor-derived factors educate macrophages to become the alternatively activated M2 type that possess protumorigenic activities such as enhancing angiogenesis, tissue remodeling and immune suppression (13,23). We again turned to a cell culture system to parse out the molecular crosstalk between tumor cells and macrophages. Treating Myc-CaP tumor cells alone with MDV3100 did not appreciably alter the expression of protumorigenic genes vascular endothelial growth factor A (Vegf-a), matrix metalloproteinase 9 (Mmp-9) or arginase 1 (Arg-1) (Figure 3a). However, it is clear that ABT can enhance the tumor cells’ expression of M2-promoting cytokines such as IL-13 and IL-10, albeit another M2 cytokine IL-4 was unaltered (16) (Figure 3b). In a binary tumor cell-macrophage co-culture system we observed a dramatic increase in the expression of VEGF-A, MMP-9, Arg-1 (Figure 3c) and of M2 cytokines IL- 10 and CSF-1, and a reduction in the pro-inflammatory M1 cytokine IL-12 (Figure 3d). These protumorigenic and M2-gene expression changes were not observed in ABT of macrophages alone (Figure 3c–d). In parallel, MDV3100 treatment of tumor bearing mice resulted in a significant increase in Vegf-a, Mmp-9, and Arg-1 expression (Figure 3e) and increased blood vessel density (Figure 3f) in MDV3100-treated tumor compared to control. A further indication that TAMs in the castrated tumors possess more protumorigenic M2-phenotype is that they displayed much lower levels of MHCII expression, consistent with an immunosuppressive state, than those in untreated tumors (Supplemental Figure 2) (27).
Figure 3. ABT promotes alternative activation of macrophages.
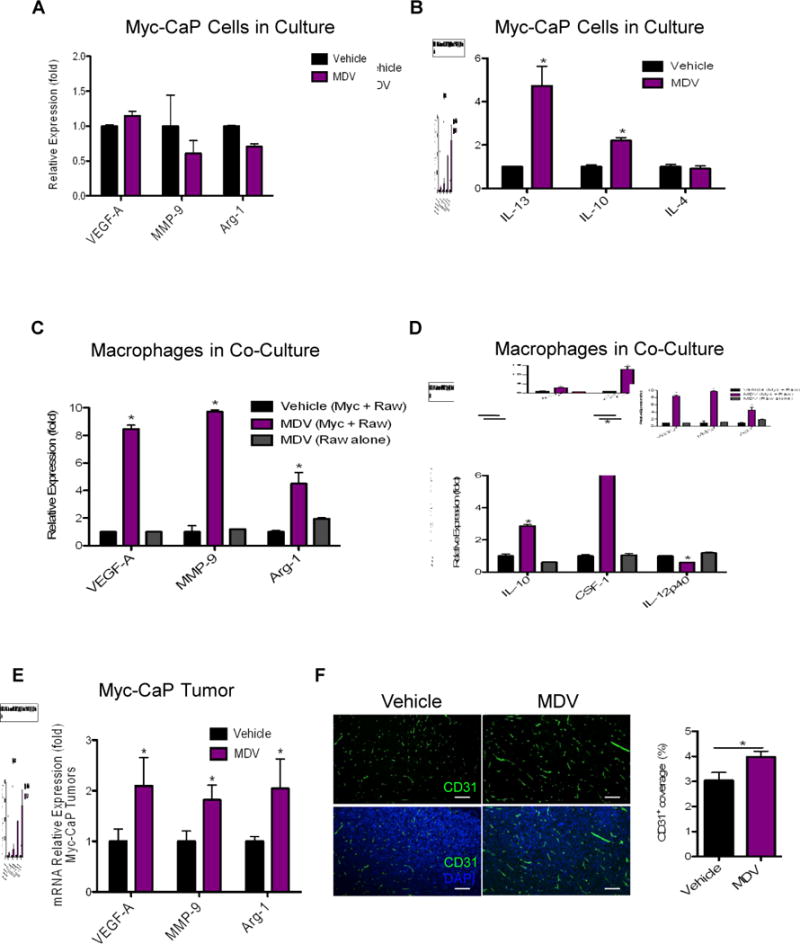
A-B) Relative gene expression in Myc-CaP tumor cells treated with MDV3100 (10 μM) for 48 hrs, normalized to vehicle treated control. C-D) RAW264.7 macrophages were co-cultured with Myc-CaP tumor cells or grown alone and treated with vehicle or MDV3100 (10 μM) for 48 hrs. Gene expression in RAW264.7 macrophages were normalized to co-cultured and vehicle-treated macrophages. E) Relative gene expression in MDV-treated Myc-CaP tumors normalized to control vehicle-treated tumors (level=1), as analyzed by RT-PCR. (n = 6–10 per group). F) Representative images of CD31 blood vasculature (green), DAPI (blue) IHF staining of treated Myc-CaP tumors. Right graph shows the quantification of CD31 staining in F shown as percent area covered (n = 4 per group). Scaled bars represent 100 μm. *P < 0.05.
Taken together, these results from in vitro and in vivo models indicate that ABT of prostate tumor cells elicits a paracrine crosstalk through soluble cytokines with TAMs that promotes their recruitment to the tumor as well as their protumorigenic properties.
CSF1R blockade in combination with ADT lowered TAMs and systemic level of myeloid cells
Our in vitro and in vivo findings pointed to CSF-1 being a critical cytokine that modulates the activities of TAMs in ABT. Hence, a rational therapeutic strategy could be to use a CSF1R kinase inhibitor to disrupt the protumorigenic influences of TAMs. PLX3397 is a recently developed small molecule kinase inhibitor that antagonizes CSF1R (c-FMS) with IC50 of 20 nM. Its functional activity (9) and chemical composition has been reported previously (28). Furthermore, PLX3397 has been under clinical investigation for several types of cancers (29).
In this study, we specifically examined the therapeutic impact of PLX3397 in combination with ADT. Since the CSF-1/CSF1R signaling axis has been implicated in prostate cancer oncogenesis (30), we first evaluated whether the therapeutic effects of blocking this axis could be directed at the tumor cells. The Myc-CaP tumor cells express negligible levels of CSF1R but relatively high levels of CSF-1 (Supplemental Figure 3a–b). The proliferation of Myc-CaP tumor cells was unaltered after effective knockdown of CSF-1 expression by shRNA or CSF1R blockade with GW2580 (Supplemental Figure 3c–d). These results are consistent with the assertion that the potential therapeutic effect of CSF1R blockade is PCa cell-extrinsic. In the pursuit of the ADT plus PLX3397 combined therapeutic strategy, we first verified the pharmacological action of PLX3397. Tumor sections were stained with anti-phosphorylated CSF1R antibody and we found that this signaling pathway is largely restricted to TAMs within the tumor (Figure 4a). Furthermore, castration clearly increased the number of phosphorylated CSF1R+ TAMs (Figure 4a). PLX3397 treatment either alone or in combination with castration lowered the number and intensity of the phosphorylated CSF1R signal (Figure 4a), confirming the expected pharmacological activity of PLX3397.
Figure 4. CSF1R blockade effectively lowered tumoral and systemic macrophage levels.
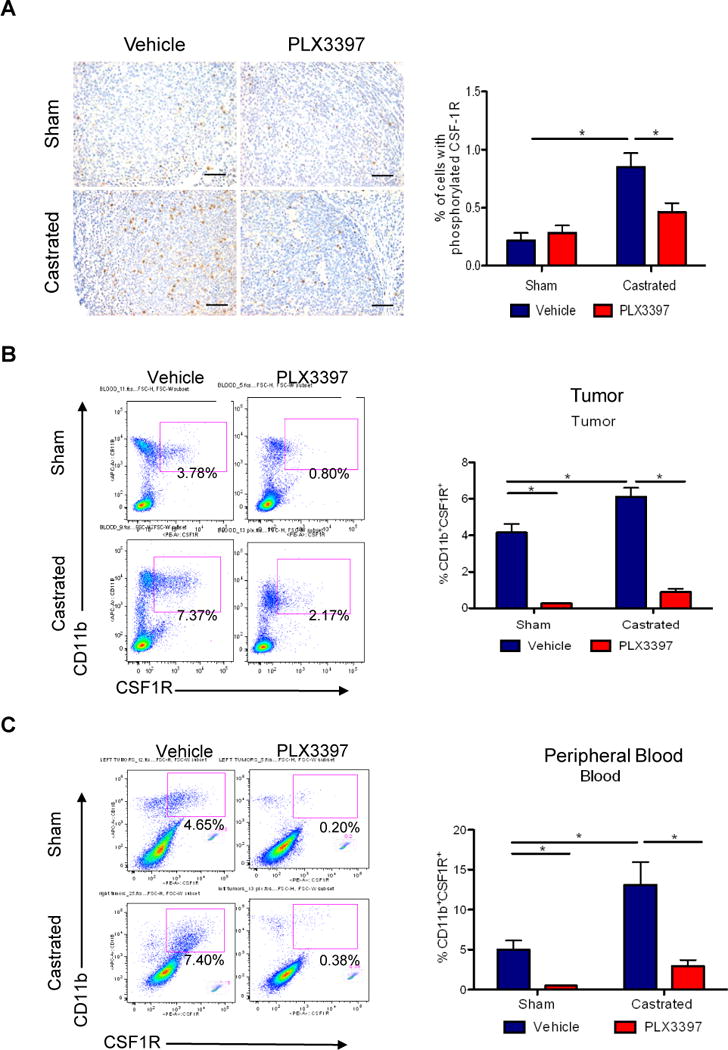
Myc-CaP tumor bearing mice received surgical castration or sham surgery when tumors reached 300–500 mm3, and then fed with control or PLX3397 chow daily for 36 days. A) Representative images of tumor sections stained (left panel) with anti-phosphorylated CSF1R (CSF1R-Tyr723) from 4 treatment groups. Scaled bars represent 100 μm. The right graph shows the quantification of IHC staining. B) Representative flow cytometry plots of B) total CD11B+CSF1R+ TAMs (left) in tumors, and C) total CD11B+CSF1R+ myeloid cells in peripheral blood, along with quantification of each (right). *P < 0.05. (n = 6–10 per group).
Detailed flow cytometric analysis of myeloid populations in the 4 treatment cohorts (sham surgery, sham + PLX3397, castration, castration + PLX3397) revealed that ADT induced a significant increase in intratumoral content of CD11b+CSF1R+ (F4/80+) macrophages from an average of 4.16 +/− 0.48 to 6.11 +/− 0.49 % of viable cells (n = 7) (Figure 4b). In turn, adding PLX3397 lowered TAM content significantly to 0.27 +/− 0.01% and 0.89 +/− 0.16% in the sham- and castration-treated tumors, respectively (Figure 4b). Based on the results of our in vitro studies (Figure 2 and 3), we anticipate the impact of tumor-directed ABT can be transmitted systemically through circulating cytokines. Specifically, the elevated CSF-1 level expressed in the tumor and secreted CSF-1 in serum (Figure 2g, h) induced by castration can account for the significant increase in the CD11b+CSF1R+ (Gr-1+) myeloid cells in the peripheral blood after castration (Figure 4c). This population was decreased effectively by PLX3397 treatment (Figure 4c). Likewise, the systemic alterations of this myeloid population in response to castration can also be observed in the spleen (Supplemental Figure 4). Of note, no overt toxicities based on body weight changes and gross observations of the mice were observed in the different PLX3397 treatment arms, up to 5 weeks in the castration + PLX3397 arm (data not shown).
CSF1R blockade abrogated the protumorigenic influences of TAMs
Parallel to the results of treatment with MDV3100 (Figure 3a–c), surgical castration also induced the expression of MMP-9, VEGF-A, and Arg-1 mRNA in Myc-CaP tumors (Figure 5a–c). Importantly, CSF1R blockade treatment via PLX3397 was able to counter the castration–induced expression of MMP-9, and VEGF-A, lowering their level to equal or below sham treated control tumors (Figure 5a, b). The expression of Arg-1 was reduced to a lesser extent (Figure 5c). Immunohistofluorescence (IHF) analysis again confirmed that ADT increased the number of TAMs (Figures 5d, left panels) as noted above by flow cytometric analyses (Figure 1e–f and 4b). Furthermore, IHF analysis revealed that F4/80+ macrophages are the predominant cell population in the tumor that expressed MMP-9 (Figure 5d, middle panels) and the proportion of TAMs expressing MMP-9 increased significantly after castration (Figure 5d middle and right panels), corroborating the gene expression findings of Figure 5a. Remarkably, PLX3397 treatment not only reversed the heightened number of TAMs recruited (Figure 4b) but also their expression of protumorigenic factors, such as MMP-9 (Figure 5a–d).
Figure 5. CSF1R Blockade lowered TAM-induced tumorigenic factors and delayed the emergence of CRPC.
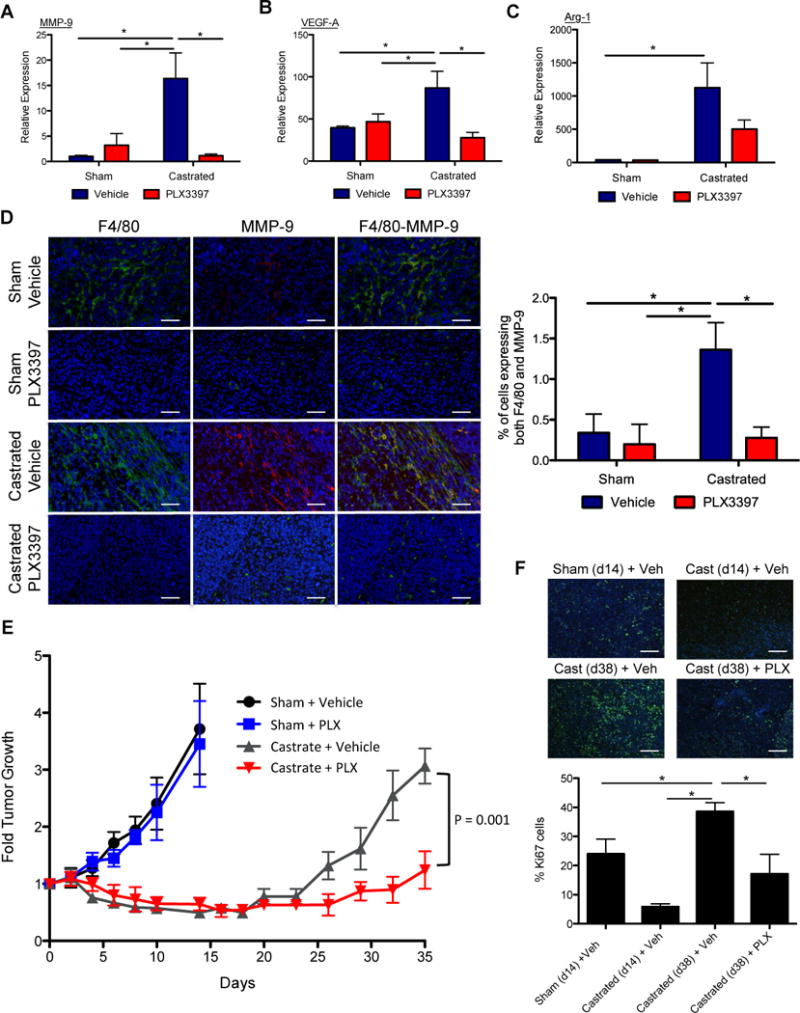
A) The impact of 4 treatment groups as noted in Figure 4 on the expression level of MMP-9, VEGF-A, and Arg-1 in the tumor. B) Representative IHF images of treated Myc-CaP tumor sections showing staining for nuclei (DAPI in blue) with F4/80 macrophages (green, left panels), or with MMP-9 (red, middle panels) singly and overlaid of all 3 stains (right panels). Right graph shows the quantification of percent of cells expressing both F4/80 and MMP-9. Scaled bars represent 100 μm. E) The impact of 4 treatments on tumor growth, expressed as fold change over start of treatment (day 0). F) The tumor proliferation rate for treated tumors was assessed by Ki67 staining. Representative IHF images from sham tumors (day 14), day 14 post-castration, and day 38 post-castration without or with PLX3397 treatment (Cast (day 38) + veh, Cast (day 38) + PLX) were shown. Ki67 (green) and DAPI (blue). Scaled bars represent 100 μm. The right graph shows the quantification of Ki67 staining for each condition, time point (total Ki67/total nuclei, n = 5). *P < 0.05. (n = 6–10 per group).
Blocking TAMs improved efficacy of ADT in murine Myc-CaP and human CWR22Rv1 prostate tumors
The protumorigenic influences of M2 TAMs induced by ABT provide a clear indication that using a CSF1R inhibitor to block TAM function could improve the efficacy of ABT. The therapeutic response of such a combined ADT and PLX3397 preclinical trial indeed support this assertion (Figure 5e). Of note, treatment with PLX3397 alone did not suppress the growth of Myc-CaP tumors compared to sham control (Figure 5e). Consistent with our previous published reports (23,31), this finding again supports our assertion that the therapeutic target of CSF1R blockade is TAMs and not tumor cells. Surgical castration alone was able to suppress Myc-CaP tumor growth. But the tumor growth rebounded approximately 20 days after surgery, paralleling the emergence of CRPC observed in the clinical setting (Figure 5e). The addition of an oral regimen of the CSF1R inhibitor PLX3397 to castration resulted in a significant delay in the onset of CRPC (Figure 5e). Examination of tumor cell proliferation by Ki67 showed that castration significantly suppressed tumor growth initially as the proportion of proliferating, Ki67-positive tumor cells in the day 14 castrated tumors is significantly lower than the control untreated (sham) tumors at the same time (Figure 5f). However, the level of CD11b+CSF1R+ TAMs in castrated tumors (6%, see Figure 1e, f) was elevated at day 14 compared to control tumors (3%). The heightened level of M2 TAMs (day 14) could augment tumor growth, accounting for the high level of proliferation observed on day 38 in the castrated group (40%). Remarkably, blocking the TAM function with PLX3397 treatment in the castrated group resulted in a dramatic lowering of Ki67 levels (17%) on day 38 (Figure 5f).
It is well documented that the plasticity of TAMs can be modulated by local tissue environment (32). Thus, we examined the impact of TAMs in an ADT setting in the human prostate xenograft CWR22Rv1, implanted orthotopically in the murine prostate gland. The CWR22Rv1 is an AR-expressing cell line that has been shown to be relatively resistant to ABT. The response to castration in the CWR22Rv1 orthotopic xenograft model is reminiscent to that in the Myc-CaP subcutaneous model, but in a contracted timeline. We observed a transient suppression of CWR22Rv1 tumor growth upon castration, as monitored by bioluminescent imaging. However, the tumor regrew quickly within about one week (Supplemental Figure 5a–b). We next examined whether inhibiting the macrophage function with PLX3397 could also delay the regrowth of CWR22Rv1 tumor in the ADT setting. Gene expression analysis and flow cytometry revealed a significant reduction in TAMs upon PLX3397 treatment (Figure 6a–b). Concurrent with macrophage depletion, expression of MMP-9 showed a reducing trend in the PLX3397 castrated compared to the vehicle-castrated group (Supplemental figure 5c). Following one week of treatment with PLX3397, there was no significant difference in tumor growth in the non-castrated group. As predicted, in the animals that received castration, PLX3397 treatment contributed to a significantly reduced regrowth of the tumor compared to castration alone (Figure 6b and Supplemental Figure 5d).
Figure 6. Blockade of TAMs extended castration response in an orthotopic xenograft model.
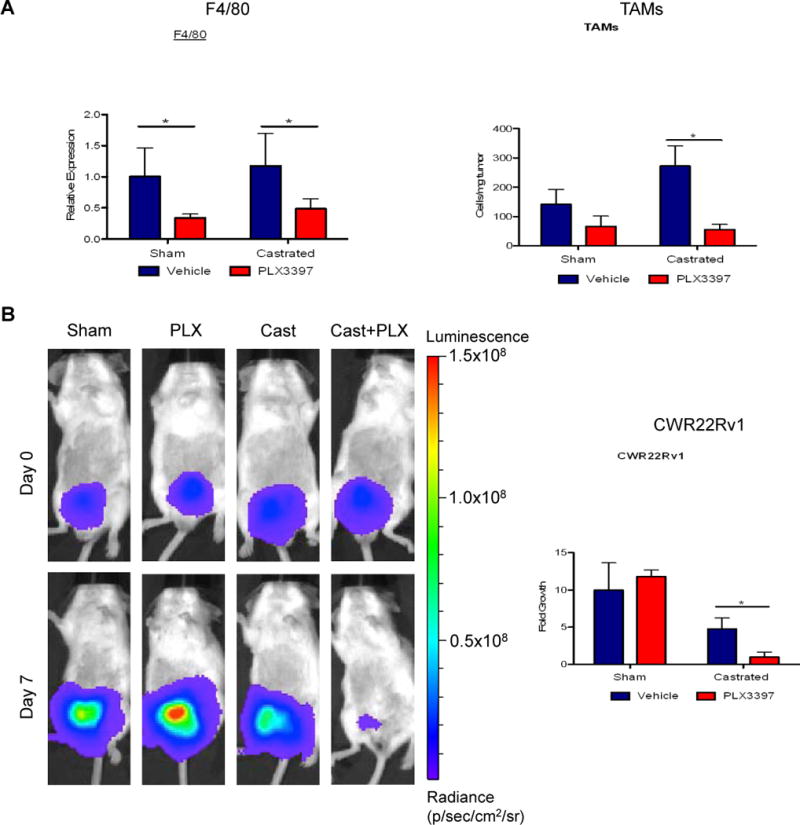
Firefly luciferase marked CWR22Rv1 tumor cells were implanted in the prostate gland of SCID/beige male mice. Tumor was allowed to establish for 2 weeks. Then the tumor bearing mice were stratified to 4 treatment groups (control, PLX3397, castration and castration+PLX3397). A) F4/80 expression analyses (left) and flow cytometric quantification of TAMs (CD45+CD11b+F4/80+, right) in the treated CWR22Rv1 tumors. B) Bioluminescent signal in orthotopic CWR22Rv1 tumors was assessed at day 0 and day 7 after castration, with representative images of animals from each group presented (left). Quantification of tumor growth by normalizing bioluminescent signal from day 7 to signal from day 0 for each mouse in the four groups (right). *P <0.05. (n = 3–4 per group).
From these results, we conclude that PLX3397 is effective at inhibiting CSF1R signaling in TAMs. In effect, CSF1R blockade serves to counter the induction of CSF-1/CSF1R signal axis set forth by ADT. Incorporating a regimen to counteract the protumorigenic actions of TAMs could be a rational approach to extend the therapeutic efficacies of conventional treatment such as ADT for prostate cancer.
Discussion
The contribution of TAMs to treatment failure is a timely issue of great interest in cancer research (32). In ABT for prostate cancer, recent preclinical and clinical studies suggested TAMs exert a negative impact on treatment response (17,18). However, these studies did not examine the causal relationship between ABT and TAMs or what protumorigenic influences the TAMs could be contributing to treatment resistance. Hence, in this study we explored the molecular signals in tumors triggered by androgen inhibition that could modulate the activity of macrophages. We observed that ABT induced prostate cancer cells to express cytokines, including CSF-1, IL-13 and IL-10, which are known to recruit and polarize macrophages towards an alternatively activated, protumorigenic state (16,33). In turn, the M2 TAMs expressed elevated levels of the VEGF-A, MMP-9 and Arg-1 genes, which can promote treatment resistance by enhancing tumoral angiogenesis, tissue-remodeling and immune suppression, respectively (16). To mitigate the negative contribution of TAMs, we blocked the CSF-1/CSF1R axis, which is critical for the function of the myeloid and macrophage lineages in particular (14). Our data here showed that the use of selective CSF1R kinase inhibitors, such as GW2580 and PLX3397, in combination with ABT was able to reverse the treatment-induced increase in the number of TAMs recruited and their protumorigenic functions, leading to more effective and prolonged tumor growth suppression than ABT alone. The results of this study indicate that ABT can induce tumor signals to recruit TAMs and modulate their functions, thus contributing to eventual treatment resistance. The central concept put forth by this study is diagramed in Figure 7.
Figure 7. Schematic diagram of ABT induced recruitment and polarization of M2-macrophages and their effects on prostate cancer progression.
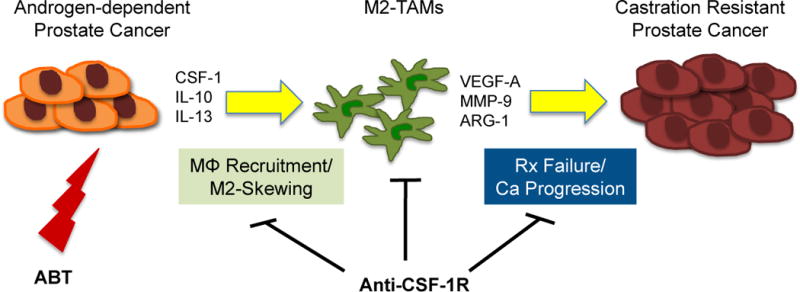
Androgen blockade therapy by chemical or physical castration and AR blockade induces expression of macrophage recruiting cytokine CSF-1 as well as M2-skewing cytokines such as IL-10 and IL-13. M2-macrophages and their tumor promoting properties can be countered by inhibitors of CSF-1/CSF1R axis, such as PLX3397 resulting in delaying treatment failure (i.e. the onset of CRPC). In effect, adding a TAM blocking treatment can improve the durability of existing ABT.
Extensive clinical experience has demonstrated that the growth and survival of prostate cancer is highly dependent on androgen and AR, even in the advanced CRPC stage of disease (34). Thus, ABT ablating either the ligand or the function of AR will continue to be a key therapeutic modality for prostate cancer. Investigations of therapeutic failure to ABT have commonly focused on tumor-intrinsic mechanisms. This study examined a tumor-extrinsic treatment bypass mechanism mediated by macrophages. As ABT is a systemic treatment, it is important to contemplate if blocking the androgen/AR axis could have a direct impact on macrophage function. A report by Lai et al. (35) showed that suppression of AR function in macrophages could promote their wound healing function. The current finding from this study is not consistent with a direct effect of ABT on macrophages. In co-culture experiments, treating a macrophage cell line (Figure 3c, d) or bone marrow derived macrophages (data not shown) directly with MDV3100 did not induce their protumorigenic phenotypes. Furthermore, macrophages either from cell lines or endogenous sources express very low levels of AR, more than 100 fold lower than the AR in our prostate tumor cell lines (data not shown). Taken together, these results suggest that the impact of ABT is directed at tumor cells, leading to the induction of paracrine signals to modulate macrophage activities.
The macrophage activities observed in this therapeutic study are consistent with their innate physiological function. It is a well-known phenomenon that dying/necrotic tumors have an increased inflammatory response. Dying tumor cells secrete cytokines that alert the immune system to respond as they would to a wound or injury (36). Macrophages are one of the first responders to a wound, and they function to remove debris and promote angiogenesis and tissue remodeling as part of the wound healing process (37). It is reasonable to postulate that the synchronous induction of cell damage in the cancer therapeutic setting would be parallel to a tissue wounding process and incites the injury responses to recruit macrophages. We demonstrated that CSF-1 is one of the key macrophage recruitment signals produced by damaged tumor cells in ABT (this study), anti-angiogenesis therapy (23), radiation therapy (RT) (31), and adoptive cell transfer immunotherapy (38). We have further characterized that the DNA damage induced by RT activates Abelson murine leukemia viral oncogene homolog 1 (ABL1) kinase and promotes it to upregulate Csf1 gene transcription (31) We are mindful that CSF-1 is just one of numerous chemokines that have been reported to modulate TAMs’ recruitment and function in different tumor types and therapeutic settings (32). Specifically, chemokine (C-C motif) ligand 2 (CCL-2, or MCP-1), and chemokine (C-X-C motif) ligand 12 (CXCL12, or SDF-1) were shown to influence prostate cancer progression and metastasis (39,40). However, in the Myc-CaP tumor model, the level of MCP-1 and SDF-1 is negligible and unchanged after ABT. This result would suggest they are not likely to be the dominant factor involved in ABT-mediated macrophage recruitment in this model. Our prior studies have also demonstrated that CSF-1 is a relevant and key macrophage recruitment and modulating factor in anti-angiogenesis and RT settings of prostate cancer (23,31).
To fully understand the mechanism of macrophage induced treatment resistance, it is instrumental to consider the likely protumorigenic signals emanating from the macrophages. In the context of ABT, an earlier study by Zhu et al. reported that macrophages and IL1β secreted by macrophages can promote an antagonist to agonist conversion of bicalutamide by modulation of AR co-activators (41). However, we could not substantiate this IL1β mediated effect in our tumor models. In contrast, we found that in the context of ABT of prostate tumor models, the macrophage phenotype changes from an M1 to an M2 state. As noted above, a large volume of evidence supports that a key protumorigenic function of M2 macrophages is to promote tumor angiogenesis and tissue remodeling (12,13,16). In fact, several studies specifically implicate the proangiogenic properties of TAMs as the culprits of resistance to anti-angiogenic therapy (23,42). We observed a significant increase of VEGF-A, and MMP-9 gene expression and tumoral angiogenesis that correlated with the increase of TAMs after ABT. Furthermore, this cytotoxic therapy not only increased the number of TAMs within the tumor, but the magnitude of protumorigenic genes per macrophage (e.g. MMP-9) is also elevated. Another plausible but currently underexplored impact of TAMs is to provide growth factors that promote tumor cell growth directly. We found that the expression of several growth factors such as VEGF, CSF-1, EGF, FGF and IGF can be induced by ABT (data not shown). The TAM contribution on each of these cytokine axes and each cytokine’s impact on tumor recurrence require further detailed investigation. At this time we can only rule out CSF-1 having a tumor-directed growth effect in our models. Even though prior reports have suggested CSF-1/CSF1R axis promotes prostate tumorigenesis (30,43), we found that neither shutdown of CSF-1 expression nor CSF1R blockade suppressed Myc-CaP tumor cell growth. Collectively, we believe it is the sum of the numerous protumorigenic properties of TAMs that are promoting the treatment failure. As such, the influx of TAMs induced by ABT resulted in heightened proliferation of the tumor. This is the first report that demonstrates the protumorigenic role of TAMs as an extrinsic bypass mechanism to an important and proven therapeutic strategy, namely androgen/AR blockade therapy, against prostate cancer.
This report brings forth several additional clinical translational considerations. A wealth of evidence indicates that the tumor-infiltrating myeloid cells and macrophages foster an immunosuppressed tumor environment (12). We consistently observed that influx of TAMs resulted in an increased expression of immune suppressive genes, such as Arg-1 and IL-10 (16). Thus, it is reasonable to anticipate that high levels of TAMs could suppress the adaptive anti-tumor T-cell response. Hence, we and other investigators (44) have begun to investigate the benefits of depleting or blocking these immunosuppressive myeloid cells, especially in the context of tumor immunotherapy. Novel immunotherapeutics such as the recently FDA approved Sipuleucel-T, which focus on activating the anti-tumoral T-cell response, have began to emerge for PCa (45). It is worthy to investigate whether incorporating a TAM-blocking regimen could further augment the therapeutic efficacy of Sipuleucel-T treatment in CRPC patients (46). Furthermore, there are numerous selective CSF1R kinase inhibitors and antibodies being developed (47–49). PLX3397 is a recently developed CSF1R kinase inhibitor that is currently in clinical trials to block myeloid cells and macrophages in several types of solid tumor (46).
This study highlights TAM’s contributions to bypass an effective conventional prostate cancer therapy. Furthermore, we demonstrate that the use of selective CSF1R kinase inhibitors could be an effective means to abrogate the protumorigenic functions of TAMs and augment the therapeutic efficacy of ABT and other conventional therapies. Early results from clinical trials suggest that the CSF1R inhibitor (PLX3397) is well tolerated by cancer patients (29). These promising results should pave the way towards the development of rational therapies that not only target tumor-intrinsic growth pathways but also the tumor extrinsic treatment bypass mechanism as noted here. The long-term outlook is that these combination approaches could extend the efficacy of conventional cancer therapeutics to benefit patients with advanced prostate cancer.
Supplementary Material
Acknowledgments
We deeply appreciate the technical advice provided by Drs. Wayne Austin and David Mulholland and Ms. Helene Hovington. This project is supported by the CDMRP PCRP award W81XWH12-1-0206 to LW. JE was supported by NCI/NIH RO1CA101904-08S1 and UCLA Cota Robles fellowship. SS was supported by the CDMRP Prostate Cancer Training Award W81XWH-11-1-0505, the NIH Tumor Immunology Training Grant 5T32CA009120 and the UCLA/Caltech MSTP T32GM008042. Flow cytometry and tissue process/immunohistochemical analyses were performed by Flow Cytometry Core Facility and Translational Pathology Core Laboratory of UCLA Jonsson Comprehensive Cancer Center, which is supported by the NCI/NIH award P50CA16042.
Footnotes
Conflict of interest: The authors of this study disclose no conflicts of interest.
References
- 1.Huggins C, Stevens RE, Hodges CV. The effect of castration on advanced carcinoma of the prostate gland. JAMA Surgery. 1941;43:209–23. [Google Scholar]
- 2.El-Amm J, Aragon-Ching JB. The changing landscape in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Med Oncol. 2013;5:25–40. doi: 10.1177/1758834012458137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene. 2014 doi: 10.1038/onc.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KAT, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 6.Kung HJ. Targeting tyrosine kinases and autophagy in prostate cancer. Hormones & cancer. 2011;2:38–46. doi: 10.1007/s12672-010-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 8.Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18:1359–68. doi: 10.1038/nm.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loges S, Schmidt T, Carmeliet P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer. 2010;1:12–25. doi: 10.1177/1947601909356574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 12.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–8. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 13.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–44. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 15.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–27. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Nonomura N, Takayama H, Nakayama M, Nakai Y, Kawashima A, Mukai M, et al. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2010;107:1918–22. doi: 10.1111/j.1464-410X.2010.09804.x. [DOI] [PubMed] [Google Scholar]
- 18.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–5. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brakenhielm E, Burton JB, Johnson M, Chavarria N, Morizono K, Chen I, et al. Modulating metastasis by a lymphangiogenic switch in prostate cancer. Int J Cancer. 2007;121:2153–61. doi: 10.1002/ijc.22900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burton JB, Priceman SJ, Sung JL, Brakenhielm E, An DS, Pytowski B, et al. Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res. 2008;68:7828–37. doi: 10.1158/0008-5472.CAN-08-1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Yao JL, Zhang L, Bourne PA, Quinn AM, di Sant’Agnese PA, et al. Differential expression of interleukin-8 and its receptors in the neuroendocrine and non-neuroendocrine compartments of prostate cancer. The American journal of pathology. 2005;166:1807–15. doi: 10.1016/S0002-9440(10)62490-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Sun Y, Wu C, Magyar CE, Li X, Cheng L, et al. Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway. Endocrine Related Cancer. 2012;19:321–31. doi: 10.1530/ERC-11-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–71. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 26.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benfan Wang QL, Li Qin, Siting Zhao, Jinyan Wang, Xiaoping Chen. Transition of tumor-associated macrophages from MHC class IIhi to MHC class IIlow mediates tumor progression in mice. BMC Immunology. 2011;12:43. doi: 10.1186/1471-2172-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Food and Drug Administration [Internet] Silver Spring(MD): Orphan Drug Designations and Approvals – [5-(5-Chloro-1H-pyrrolo[2,3-b]pyridin-3-ylmethyl)-pyridin-2-yl]-(6-trifluoromethyl-pyridin-3-ylmethyl)-amine hydrochloride salt. [Cited 2014 Sept 26] Available from: http://www.accessdata.fda.gov/scripts/opdlisting/oopd/OOPD_Results_2.cfm?Index_Number=419913.
- 29.Anthony S, Puzanov I, Lin P, Nolop K, West B, Von Hof D. Pharmacodynamic activity demonstrated in phase I for PLX3397, a selective inhibitor of FMS and Kit. J Clin Oncol. 2011;29 [Google Scholar]
- 30.Ide H, Seligson DB, Memarzadeh S, Xin L, Horvath S, Dubey P, et al. Expression of colony-stimulating factor 1 receptor during prostate development and prostate cancer progression. Proc Natl Acad Sci U S A. 2002;99:14404–9. doi: 10.1073/pnas.222537099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–86. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Kim J, Modlin RL, Moy RL, Dubinett SM, McHugh T, Nickoloff BJ, et al. IL-10 production in cutaneous basal and squamous cell carcinomas. A mechanism for evading the local T cell immune response. J Immunol. 1995;155:2240–7. [PubMed] [Google Scholar]
- 34.Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clin Cancer Res. 2011;17:1649–57. doi: 10.1158/1078-0432.CCR-10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai JJ, Lai KP, Chuang KH, Chang P, Yu IC, Lin WJ, et al. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. The Journal of clinical investigation. 2009;119:3739–51. doi: 10.1172/JCI39335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 37.Van Ginderachter JA. The wound healing chronicles. Blood. 2012;120:499–500. doi: 10.1182/blood-2012-05-430660. [DOI] [PubMed] [Google Scholar]
- 38.Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF1 Receptor Improves the Anti-tumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res. 2014;74:153–61. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9:556–62. doi: 10.1593/neo.07307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–7. [PubMed] [Google Scholar]
- 41.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–29. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 42.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106:6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ide H, Hatake K, Terado Y, Tsukino H, Okegawa T, Nutahara K, et al. Serum level of macrophage colony-stimulating factor is increased in prostate cancer patients with bone metastasis. Hum Cell. 2008;21:1–6. doi: 10.1111/j.1749-0774.2007.00042.x. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava MK, Zhu L, Harris-White M, Kar UK, Huang M, Johnson MF, et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS One. 2012;7:e40677. doi: 10.1371/journal.pone.0040677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller AM, Pisa P. Tumor escape mechanisms in prostate cancer. Cancer Immunol Immunother. 2007;56:81–7. doi: 10.1007/s00262-005-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu C, Williams AK, Chalasani V, Martinez CH, Chin J. Immunotherapy for metastatic prostate cancer: where are we at with sipuleucel-T? Expert Opin Biol Ther. 2011;11:99–108. doi: 10.1517/14712598.2011.538677. [DOI] [PubMed] [Google Scholar]
- 47.Ohno H, Uemura Y, Murooka H, Takanashi H, Tokieda T, Ohzeki Y, et al. The orally- active and selective c- Fms tyrosine kinase inhibitor Ki20227 inhibits disease progression in a collagen- induced arthritis mouse model. European journal of immunology. 2008;38:283–91. doi: 10.1002/eji.200737199. [DOI] [PubMed] [Google Scholar]
- 48.Manthey CL, Johnson DL, Illig CR, Tuman RW, Zhou Z, Baker JF, et al. JNJ-28312141, a novel orally active colony-stimulating factor-1 receptor/FMS-related receptor tyrosine kinase-3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Mol Cancer Ther. 2009;8:3151–61. doi: 10.1158/1535-7163.MCT-09-0255. [DOI] [PubMed] [Google Scholar]
- 49.MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–63. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.