

Abstract
The phosphatases PTEN and INPP4B have been proposed to act as tumor suppressors by antagonizing PI3K/AKT signaling, and are frequently dysregulated in human cancer. While PTEN has been extensively studied, little is known about the underlying mechanisms by which INPP4B exerts its tumor suppressive function and its role in tumorigenesis in vivo. Here, we show that a partial or complete loss of Inpp4b morphs benign thyroid adenoma lesions in Pten heterozygous mice into lethal and metastatic follicular-like thyroid cancer (FTC). Importantly, analyses of human thyroid cancer cell lines and specimens reveal INPP4B downregulation in FTC. Mechanistically, we find that INPP4B, but not PTEN, is enriched in the early endosomes of thyroid cancer cells, where it selectively inhibits AKT2 activation and in turn tumor proliferation and anchorage-independent growth. We therefore identify INPP4B as a novel tumor suppressor in FTC oncogenesis and metastasis through localized regulation of PI3K/AKT pathway at the endosomes.
Keywords: Cancer, metastasis, genetics, INPP4B, endosome, PI3K/AKT, thyroid
Introduction
The phosphoinositide 3-kinase (PI3K)/AKT signaling pathway modulates important biological processes such as cell cycle, survival, metabolism and motility, which are often disrupted in cancer (1). Hyperactivation of the PI3K/AKT pathway leads to an accumulation of phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3), which in turn increases the recruitment and activation of protein kinase AKT at the cytoplasmic membrane (1), a key mediator of the oncogenic effects of enhanced PI3K signaling.
The AKT protein kinase family is made up of three highly homologous isoforms, namely AKT1, AKT2 and AKT3 (2). Despite similarities in sequence and regulation, studies reveal isoform-specific functions in cancer progression. In the context of breast cancer, overexpression of AKT2 promotes metastasis (3), while AKT1 suppresses metastasis (4, 5). The distinct functions of the AKT isoforms could be explained, at least in part, through the association of the AKT isoforms with distinct organelles or subcellular compartments (2).
The tumor suppressor gene PTEN (the phosphatase and tensin homolog), encoding a lipid-phosphatase, dephosphorylates PI(3,4,5)P3 to PI(4,5)P2 to antagonize PI3K/AKT signaling. To study the effects of Pten loss in vivo, we previously generated mouse models with constitutive and conditional Pten loss (6). We found that homozygous loss of Pten results in embryonic lethality, while Pten heterozygous (Pten+/−) mice develop tumors of the breast, endometrium, prostate, adrenal, pituitary and lymphoma (7). More recently, INPP4B (inositol polyphosphate-phosphatase type II), another dual specificity and lipid phosphatase, has emerged as a putative tumor suppressor in the suppression of the PI3K/AKT signaling pathway. INPP4B was initially identified as an anchorage independent growth suppressor in a shRNA-mediated genetic screen performed in HMEC cells (8), and was found to inhibit PI3K signaling and to display tumor suppressive activity in breast tumor cell lines (9). In agreement with these findings, INPP4B expression was found to be reduced in basal-like breast cancers (9, 10), melanoma (11), nasopharyngeal carcinoma (12), and prostate cancer (13). Furthermore, expression-profiling analyses revealed that INPP4B mRNA expression is reduced in up to 47% of metastatic prostate cancer cases (14), implicating the potential role of INPP4B in metastatic progression.
INPP4B converts PI(3,4)P2 to PI(3)P and because direct interaction of PI(3,4)P2 with the pleckstrin homology (PH) domain of AKT is required for membrane recruitment and full activation of AKT (15), INPP4B, like PTEN, is anticipated to act as a tumor suppressor by antagonizing PI3K/AKT signaling (9). However, unlike PTEN, the underlying molecular mechanisms by which INPP4B exerts its tumor suppressive function are poorly understood. Additionally it is not known whether INPP4B acts as a tumor suppressor in vivo.
We therefore sought to investigate the tumor suppressive functions of INPP4B in vivo in Knock-out (KO) mouse models. Surprisingly, we found that INPP4B exerts a specific role in the suppression of thyroid tumorigenesis and metastasis in vivo through the inhibition of PI3K-C2α mediated AKT2 activation at endosomes.
Results
Inpp4b knockout (Inpp4b−/−) mice do not develop the tumorigenic phenotype of Pten haploinsufficient mice
To determine the in vivo tumor suppressive function of INPP4B, we took advantage of Inpp4b knockout mice (Inpp4b−/−) generated by using a homologous recombination targeting strategy in which the conditional targeting vector was constructed to delete exon 21 of the mouse Inpp4b gene, which encodes the phosphatase catalytic domain (Fig. 1A-B). Unlike Pten−/− mice, Inpp4b−/− mice were viable and born in accordance to Mendelian frequencies (Fig. 1C). Furthermore, Inpp4b+/− and Inpp4b−/− mice did not develop any of the tumors or the lymphoproliferative disease characteristic of Pten+/− mice in a 16-24 months follow-up (16). Aggressive and fatal, albeit sporadic, histiosarcomas were observed after a very long latency in Inpp4b−/− mice (data not shown). These results demonstrate unequivocally that the role of PTEN and INPP4B in tumorigenesis could be very distinct.
Figure 1. Generation and Characterization of Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice.
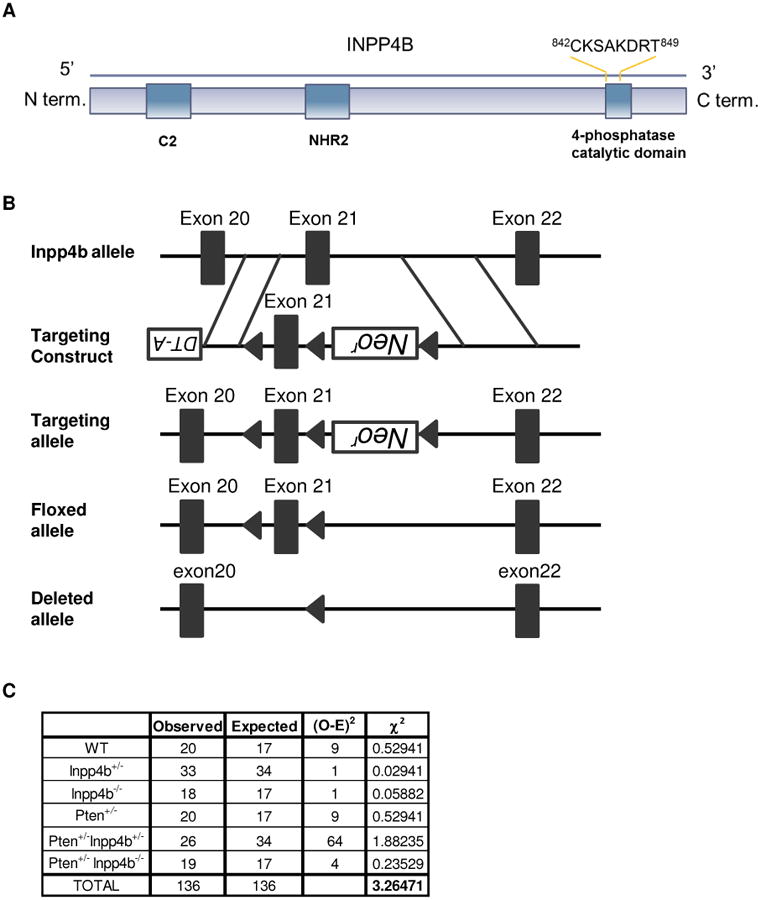
A. Diagram representing the structure of INPP4B, which contains an N-terminus C2 domain and a C-terminus phosphatase domain harboring the phosphatase catalytic motif CX5R. B. Schematic map of the WT Inpp4b locus (top), targeting vector (upper middle) and predicted targeted allele (lower middle) and knockout allele (bottom). C. Table depicting the observed versus the expected numbers of mice of the respective genotypes from a Pten+/−Inpp4b+/− and Inpp4b+/− cross. These values gave a χ2 of 3.26, which is lower than the critical value of 11.070 which would yield an α=0.05. Thus, we conclude that the mutants were born following Mendelian frequencies.
Loss of Inpp4b in a Pten heterozygous background leads to metastatic follicular-like thyroid carcinoma
We next sought to determine if loss of Inpp4b cooperated with Pten loss to promote tumor progression. We hypothesized that loss of Inpp4b would accelerate overall tumor progression in Pten+/− mice, keeping with their reported epistatic relationship in the suppression of PI3K/AKT signaling. To this end, we crossed Pten+/− mice with Inpp4b−/− mice. The resulting Pten+/−Inpp4b+/− mice were then crossed with Inpp4b+/− littermates to generate wild type, Pten+/−, Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice (Suppl. Fig. 1A). These mice were, once again, viable and born following the expected Mendelian frequencies (Fig. 1C). The lack of embryonic lethality in any of the two compound Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− genetic make-ups further underscore the fact that PTEN and INPP4B might exert distinct roles in signaling since the progressive PI3K/AKT elevation should result in embryonic lethality as previously reported (6).
However, Pten+/−Inpp4b−/− mice did not survive beyond 5-6 months of age, while the majority Pten+/−Inpp4b+/− mice died between 8 and 14 months of age (Fig. 2A). Gross anatomical analyses showed that the Pten+/−Inpp4b−/− mice developed large multinodular goiters (Fig. 2B). These mice died, or were euthanized, after developing compressive airway and esophageal obstruction as a consequence of the mass effect from thyroid enlargement. Histopathological analyses revealed that most of the thyroid tumors developed in the Pten+/−Inpp4b−/− mice had variable degrees of encapsulation (Fig. 2C and 2D, top left), microfollicular architecture (Fig. 2C, left middle panel) and many showed impressive vascular invasion closely resembling follicular thyroid carcinoma (FTC) in human. High-grade features were commonly seen, including significant mitotic activity and necrosis, while some portion of the tumors showed nuclear features of follicular variant papillary thyroid carcinoma (FV-PTC), such as nuclear contour irregularity, nuclear grooves, intranuclear pseudoinclusions and chromatin pallor (Fig. 2C, right panels). Collectively, the majority of tumors in Pten+/−Inpp4b−/− mice displayed histological and pathological features of FTC and FV-PTC in human (Fig. 2C and 2D, top). Importantly, 50% of Pten+/−Inpp4b−/− mice developed diffuse pulmonary metastases (Fig. 2D, top right). These metastases had the histologic appearance of thyroid tissue with follicular architecture, colloid, and also stained positive for thyroglobulin, a thyroid specific marker (Fig. 2D, middle panel). Necropsy did not reveal metastases in other regional and distant sites. Akt activation was observed by immunohistochemistry in both the thyroid tumors and the metastases (Fig. 2D, bottom panel). Serum TSH levels of Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice did not differ significantly from that in Pten+/− mice (Suppl. Fig. 1B). Additionally, we examined the thyroids from a cohort of Pten+/−Inpp4b+/− mice. Histopathological analyses revealed that Pten+/−Inpp4b+/− mice developed benign goiter and FVPTC/FTC, and presented with pulmonary metastases of thyroid carcinoma at a similar penetrance to Pten+/−Inpp4b−/− mice (Suppl. Fig. 1C). Furthermore, Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice did not display accelerated tumorigenesis in other tissues as compared to Pten+/− mice by 5-8 months follow-up (data not shown). However, we did note a non-significant increased incidence of breast adenocarcinoma in Pten+/−Inpp4b+/− mice (p=0.16, 37.5% in Pten+/−Inpp4b+/− vs 15.5% in Pten+/− mice). Therefore, Inpp4b loss cooperates with Pten haploinsufficiency to promote thyroid cancer progression and metastasis.
Figure 2. Loss of Inpp4b in Pten+/− mice leads to follicular thyroid carcinoma.
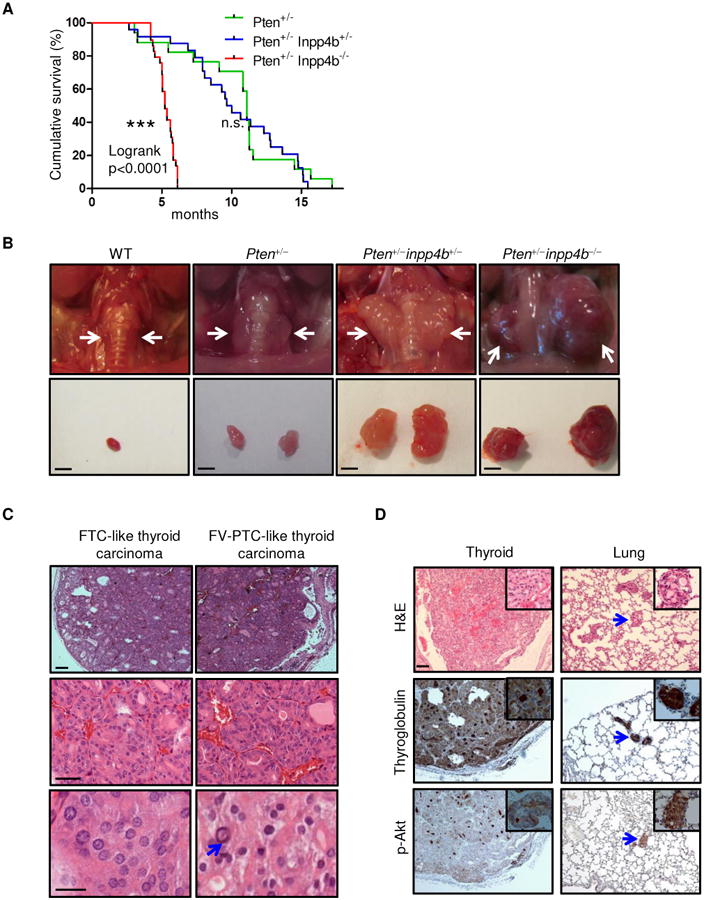
A. Kaplan-Meier survival curve of Pten+/−, Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice. B. Top panel: gross anatomy of representative thyroids from mice of the indicated genotypes. White arrows point to the location of the thyroid. Bottom panel: dissected thyroids from mice of the indicated genotypes; Scale bar, 2mm. C. Left panel: H&E staining of a follicular thyroid carcinoma (FTC)-like thyroid tumor which is thinly encapsulated with microfollicular architecture. Right panel: H&E staining of a follicular variant of papillary thyroid carcinoma (FV-PTC)-like thyroid tumor. Middle and bottom panels highlight nuclear atypia characteristic of FV-PTC, including intranuclear pseudoinclusions (arrow). Scale bars, top and middle panels: 100μm, bottom panel: 20 μm, D. Top panel: H&E staining of thyroid tumor and lung metastases from Pten+/−Inpp4b−/− mice; middle & bottom panel: thyroglobulin and p-Akt (Ser473) staining. Scale bar, 100μm. Insets show thyroid cancer cells. Blue arrows point to the location of the metastases in the lungs.
Human follicular thyroid cancer cell lines and tissues display low INPP4B expression
To assess the relevance of our mouse model findings to human FTC, we evaluated the status of INPP4B in human thyroid cancer cells. To this end we utilized seven thyroid cell lines, namely Nthy-Ori 3, Htori (SV40-immortalized primary thyroid follicular epithelial cells), FTC133, FTC236, FTC238 (follicular thyroid carcinoma), TPC1 (thyroid papillary carcinoma), and 8505C (anaplastic carcinoma). We first determined the relative expression levels of INPP4B in these thyroid cell lines. We observed very low expression of INPP4B at both protein (Fig. 3A, Suppl. Fig. 2) and mRNA transcript levels (Fig. 3B) in the FTC cell lines FTC133, FTC236 and FTC238 compared to SV40-immortalized human thyroid follicular cells Nthy-Ori-3 and Htori or the TPC1 thyroid cancer cell line. We also noted that PTEN protein expression was not detectable in the FTC cell lines (Fig. 3A). A search using the Broad CCLE database revealed that the FTC cell lines harbor the PTEN R130* nonsense mutation. This concomitant loss of PTEN and INPP4B in our human FTC cell lines is faithfully modelled by our Pten+/−Inpp4b−/− mice. Finally, we observed an increase in the activation of AKT in these cell lines, as indicated by higher levels of AKT phosphorylation at both Threonine 308 and Serine 473 residues (Fig. 3C).
Figure 3. INPP4B expression is reduced in human follicular thyroid cell lines and human surgical specimens.
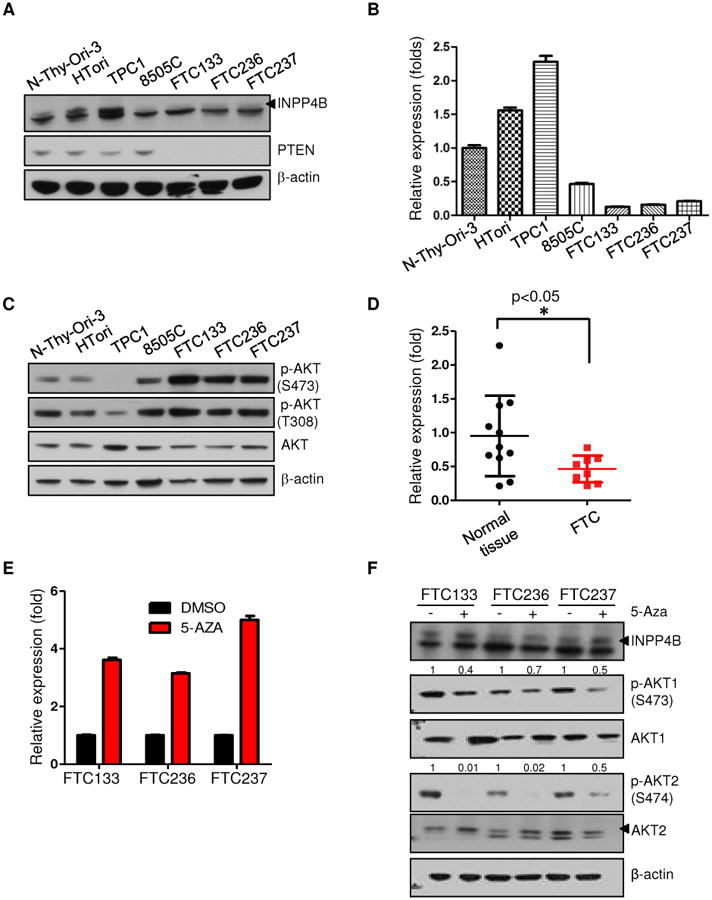
A. Western blot analysis of thyroid cancer cell lines for INPP4B and PTEN expression. Arrow indicates specific band. The arrowhead indicates the specific band of INPP4B protein. B. RT-qPCR analysis of thyroid cancer cell lines for INPP4B transcript levels. C. Western blot analysis of thyroid cancer lines for AKT activation on both Serine 473 and Threonine 308 residues. D. RT-qPCR analysis of INPP4B in unmatched normal versus FTC patient tumor samples. E-F. Thyroid cancer cell lines were treated with 3uM 5-Aza-2′-deoxycytidine for 5 days. Panel shows transcript (E) and protein analysis of INPP4B expression and AKT activation levels in DMSO versus 5-Aza treated FTC cells. (F). Arrows indicate specific band (see also Methods). AKT2 antibody used is (5239).
Importantly, INPP4B transcript expression was significantly downregulated in human FTC samples when compared to normal tissue samples (Fig. 3D). Because of the infrequency of INPP4B deletions or mutations in human cancers, we further hypothesized that its loss in FTC cell lines might be due to aberrant gene methylation. Indeed, treatment of these cell lines with 5-Aza-2′-deoxycytidine (5-Aza), which inhibits DNA methylation, increased INPP4B expression approximately 4-fold (Fig. 3E). Upregulation of INPP4B by 5-Aza decreased activation of both AKT1 and AKT2 (Serine 473 and Serine 474 respectively), (Fig. 3F) with a much greater effect on p-AKT2 than on p-AKT1 (Fig. 3F). However, bisulfite sequencing indicated that this upregulation is potentially indirect (Suppl. Fig. S3), mediated via the upregulation of yet to be defined transcription factors. Overall, the downregulation of INPP4B in human FTC strongly supports its tumor suppressive role in this aggressive tumor type of the thyroid.
Loss of Inpp4b increases Akt activation
We previously demonstrated that INPP4B knock down enhanced AKT activation (9). To gain insight into the mechanisms by which Inpp4b might promote thyroid tumor development, we analyzed thyroid tumor lysates from Pten+/−, Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice for Akt activation via Western blotting. In agreement with previous findings, tumors from Pten+/−Inpp4b−/− mice showed higher levels of Akt phosphorylation (monitored through p-Akt1 Serine 473 and p-Akt2 Serine 474) when compared to thyroid tumors obtained from Pten+/− or Pten+/−Inpp4b+/− mice (Fig. 4A). We also isolated primary mouse embryonic fibroblasts (MEFs) of all genetic combinations – wildtype, Inpp4bflox/flox; Inpp4bflox/+; Pten+/−, Pten+/−Inpp4b+/−, and Pten+/−Inpp4b−/−. We found that MEFs with Inpp4b deletion displayed increased Akt activation, monitored through the phosphorylation of both Serine 473 and Threonine 308 at different time points after serum stimulation (Suppl. Fig. S4). In addition, although Pten+/−, Pten+/−Inpp4b+/−, and Pten+/−Inpp4b−/− MEFs did not display much higher initial Akt activation upon serum starvation-restimulation (Fig. 4B, t=5 mins and 15 mins), there was a prolonged Akt-1 and Akt2 activation in Pten+/−Inpp4b+/−, and Pten+/−Inpp4b−/− MEFs when compared to Pten+/− MEFs (Fig. 4B, t=30 mins). Furthermore, we found that human thyroid cancer cell lines with lower INPP4B displayed higher levels of AKT phosphorylation (Fig. 3C and 4C). Notably, both AKT1 and AKT2 were activated to a similar extent in total cell lysates (Fig. 4A, 4B and 4C). Therefore, loss of both lipid phosphatases, INPP4B and PTEN, resulted in an additive activation of AKT in thyroid tissue, MEFs and cancer cell lines.
Figure 4. Loss of Inpp4b in Pten+/− cells leads to increased Akt activation.
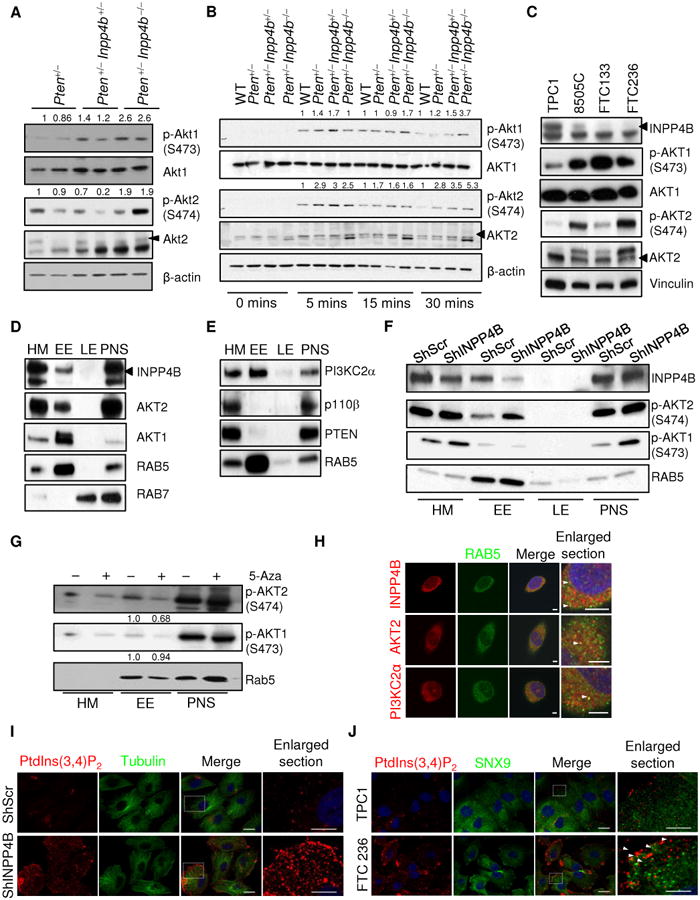
A-B. Western blot analysis of lysates from Pten+/− (n=3; ∼10-11months), Pten+/−Inpp4b+/− (n=2; ∼6-8months) and Pten+/−Inpp4b−/− (n=3; ∼6 months) thyroid tissues (A), lysates from immortalized MEFs after serum starvation-restimulation. The MEFs were stimulated with serum for the indicated amount of time. (B). C. Western blot analysis of total lysate from different thyroid cancer cells. Please note that the AKT2 antibody used in A-B (5239) is different from C (3063), resulting in the appearance of different unspecific bands. Arrows indicate specific band (see also Methods). D-E. Western blot analysis of cell fractions from TPC1 cells. HM: cytosolic/heavy membranes; EE: early endosome: LE: late endosome; PNS: postnuclear supernatant. Arrow indicates specific band. AKT2 antibody used is (3063). F. Western blot analysis of phosphorylated Akt1 and phosphorylated Akt2 in different cell fractions derived from TPC1 cells infected with either a non-targeting shRNA or an shRNA that targets INPP4B. G. Western blot analysis of phosphorylated Akt1 and phosphorylated Akt2 in different cell fractions derived from FTC236 cells treated with either DMSO or 3uM 5-Aza for 5days.
H. Immunofluorescence of PI(3,4)P2 and tubulin in TPC1 cells infected with either a non-targeting shRNA or an shRNA that targets INPP4B. Scale bars, 20 μm. I. Immunofluorescence of INPP4B, AKT2, PI3K-C2α and RAB5 in TPC1 cells. Scale bars, 5μm. J. Immunofluorescence of PI(3,4)P2 and SNX9 in TPC1 and FTC236 cells. Scale bars, 20 μm.
INPP4B suppresses PI3K-C2α mediated AKT2 activation at early endosomes in thyroid cancer cells
The observation that complete inactivation of Pten in mouse thyroid results mainly in follicular adenoma within 12 months of age (17) and that Inpp4b loss does not accelerate the entire tumor spectrum of Pten+/− mice raised the possibility that loss of Inpp4b specifically cooperates with Pten loss for tumor progression and metastasis in thyroid, not through a generic elevation of PI3K/AKT signaling, but potentially by deregulating the PI3K pathway in a more selective manner. Previous studies have shown that Akt2 deficiency had little-to-no effect on the tumor spectrum in Pten+/− mice, and only specifically significantly decreased the incidence of thyroid tumors in Pten+/− mice (18), suggesting that AKT2 might play an oncogenic role in thyroid cancer. Recent studies have shown that AKT2 phosphorylates its substrates on both endosomes and the plasma membrane (19). Strikingly, subcellular fractionation experiments showed that INPP4B, but not PTEN, was expressed in the Rab5-positive early endosomal (EE) fraction together with AKT2 in thyroid cancer cells (Fig. 4D and 4E). Although AKT1 was also localized in the EE fraction (Fig. 4D), surprisingly, knockdown of INPP4B selectively activated AKT2, but not AKT1 in the EE (Fig. 4F). Consistent with this result, thyroid cancer cell lines with lower INPP4B expression display higher endosomal AKT2 activation (Suppl. Fig. S5A). Furthermore, increasing INPP4B expression with 5-Aza in FTC236 cells resulted in a greater decrease in p-AKT2 in the EE fraction compared to p-AKT1 (Fig. 4G). Collectively, these results lend support to the selective activation of AKT2 at the early endosomes by INPP4B.
In line with class II PI3K kinase a (PI3K-C2α) producing PI(3,4)P2 and being selectively activated in endocytosis as well as endocytic recycling (20, 21), we also found PI3K-C2α expressed in the thyroid cancer cells and enriched in EE fraction, along with INPP4B and AKT2 (Fig. 4E). The association of INPP4B, AKT2 and PI3K-C2α with EE was also confirmed by immunofluorescence, in which INPP4B, AKT2 and PI3K-C2α were co-localized with RAB5-positive punctate structures in the cells (Fig. 4H). Notably, INPP4B loss was associated with increased abundance of PI(3,4)P2 (Fig. 4I and 4J), but not PI(3,4,5)P3 (Suppl. Fig. S5B). Consequently, there was an accumulation of the PI(3,4)P2-binding protein SNX9 in vesicles near the plasma membrane (Fig. 4J). Taken together, these results suggest that INPP4B negatively regulates PI3K-C2α signaling at the EE. Furthermore, loss of PI3K-C2α decreased AKT2 activation to a greater extent than that of AKT1 in total cell lysates (Suppl. Fig. S6A and S6B) and significantly inhibited cell proliferation in FTC 236 cells (Suppl. Fig. S6C), underscoring the functional relevance of enhanced PI3K-C2α signaling in cells with lower INPP4B. Taken together, our results strongly suggests that INPP4B inhibits PI3K-C2α mediated AKT2 activation in the early endosomes of thyroid cancer cells, even though they obviously do not rule out the possible involvement of other PI3K isoforms in AKT activation at the EE.
Knockdown of INPP4B in thyroid cell lines provide an advantage for anchorage independent growth
To assess INPP4B dependent cellular processes in vitro, we next conducted cell proliferation and soft agar colony formation assays in two of the non-FTC thyroid cell lines, namely TPC1 and 8505C, in which we stably knocked down INPP4B (Fig. 5A and 5B). We found that the knockdown of INPP4B did not confer a growth advantage for thyroid cell lines in full or 1% serum growth conditions, but did in 5% serum growth conditions (Fig. 5C and Suppl. Fig. S7A), suggesting that under specific conditions of nutrients or growth factor amounts, INPP4B loss determines a growth advantage in thyroid cell lines, mirroring the in vivo phenotype. In addition, INPP4B knock down did not result in any change in TPC1 cell morphology, nor did it alter the morphology of Pten+/−Inpp4b−/− MEFs compared to Pten+/− MEFs (Suppl. Fig. S7B and S7C). Furthermore, INPP4B loss did not alter the distribution or arrangement of tubulin in the cytoskeleton of these cells (Suppl. Fig. S7B and S7C). Instead, INPP4B loss provided a marked advantage for anchorage independent growth, a functional indicator of tumorigenicity and invasiveness (Fig. 5D and 5E).
Figure 5. Knockdown of INPP4B in thyroid cancer cells provides an advantage for anchorage independent growth.
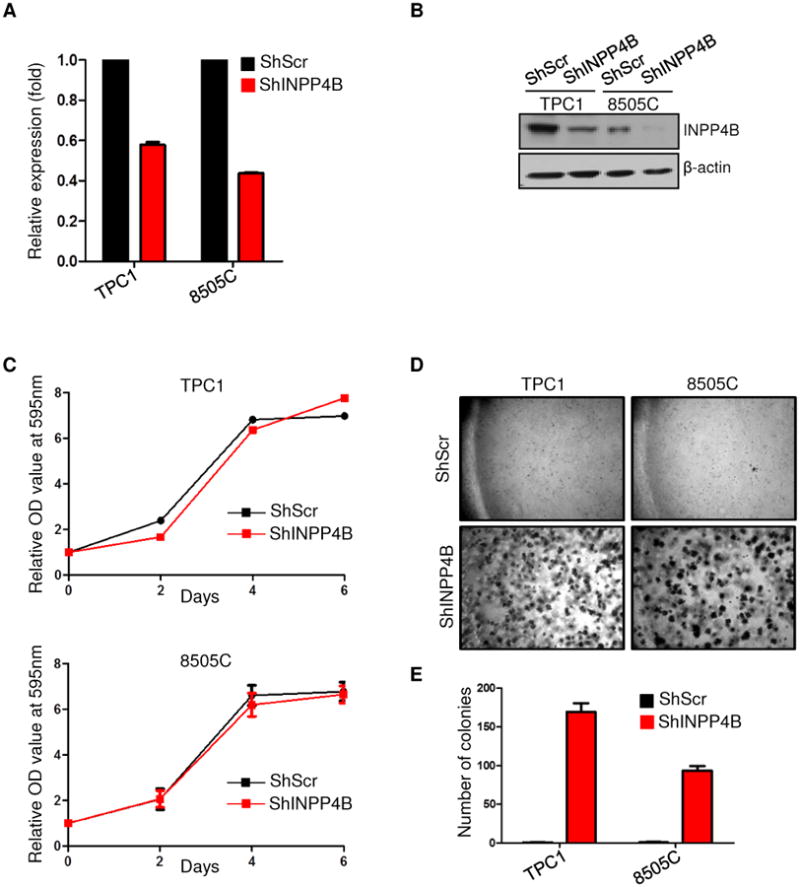
A. RT-qPCR analysis of INPP4B upon shRNA-mediated knockdown. B. Western blot analysis of INPP4B levels in TPC1 and 8505C cell lines after shRNA-mediated knockdown. C. Proliferation of TPC1 and 8505C cell lines infected with either a non-targeting shRNA or a shRNA that targets INPP4B. Cells were cultured in complete media (10% serum), stained with crystal violet, and lysed. Absorbance was measured at OD595nm. D-E. Soft agar colony formation assay of cells infected with either a non-targeting shRNA or an shRNA which targets INPP4B. Cells were grown in soft agar for 2 weeks before being photographed (D) and the number of colonies was then counted (E).
Discussion
This study allowed us to reach three important conclusions:
First, INPP4B is a novel tumor suppressor in FTC. Through our mouse model, we demonstrated that either a partial or complete loss of Inpp4b in a Pten heterozygous background accelerates thyroid carcinoma progression, resulting in metastatic disease that recapitulates the hematogenous pulmonary metastases characteristic of advanced FTC in humans. The evidence that INPP4B is a relevant tumor suppressor in FTC is further supported by the observation that INPP4B expression is markedly reduced in human FTC, as compared to a normal thyroid and other subtypes of thyroid cancer.
Our findings lend further support to the notion that PI3K-AKT activation plays a central role in FTC oncogenesis. Multiple lines of evidence corroborate this notion: 1) AKT activation and expression is higher in FTC as compared to normal tissues and other thyroid tumors (22), 2) Cowden syndrome patients, with germline mutations in PTEN, have an increased incidence of FTC (23), 3) transgenic mice engineered to hyperactivate PI3K/AKT form FTCs (24, 25), and 4) in a mouse model of FTC, metastatic potential is Akt-dependent (26). This evidence underscores the importance of AKT hyperactivation in the initiation and progression of FTC, and also suggests that the pathogenesis of FTC is distinct from other forms of well-differentiated thyroid carcinoma, which are driven primarily via activation of the MAPK signaling pathway. Nevertheless, mutations in the RAS pathway and PAX8/PPARγ rearrangements are also found in FTC (27). While the specific pathogenic mechanisms contributed by these alterations remain unclear, there are indications that they too converge on the PI3K-AKT pathway (28). Therefore, it will be therefore interesting to understand in future studies whether these represent cooperative or mutually exclusive events.
Second, we observed that INPP4B is not solely epistatic to PTEN. PTEN and INPP4B are generally thought to be cooperative phosphatases that proximally regulate PI3K at the lipid level. On this basis, we anticipated that Inpp4b−/− mice would have a phenotype reminiscent of Pten+/− mice with increased susceptibility to epithelial tumors. Surprisingly, Inpp4b−/− mice were tumor free and had a tumor free survival of 16-24 months. In addition, crossing Inpp4b−/− mice with Pten+/− mice did not accelerate the entire tumor spectrum of Pten+/− mice by 5-8 months follow-up. Rather, we only observed a significant acceleration of thyroid tumorigenesis. Due to the early mortality that occurred in the Pten+/−Inpp4b−/−, further studies in conditional Pten and Inpp4b knockout mice will be needed in order to determine the potential cooperative effect between Pten and Inpp4b on tumor suppression in other tissues.
Despite the importance of PI3K-AKT signaling in the pathogenesis of FTCs, Pten+/− mice do not develop FTC. By 10 months of age, they instead develop benign adenomas that do not invade or metastasize. One might speculate that the extent of AKT activation accounts for the difference in thyroid carcinoma aggressiveness. Indeed, we observed increased Akt activation in Pten+/−Inpp4b−/− thyroids when compared to Pten+/− or Pten+/−Inpp4b+/− thyroids. However, in a previous study, we found that thyroid lesions in Pten knock-in mutant mice harboring specific lipid and phosphatase dead Pten mutations also demonstrated higher levels of Akt activation than Pten+/− thyroids similar to Pten+/−Inpp4b−/− thyroids (29). Nevertheless, only 7 – 8 % of these mice developed aggressive thyroid adenocarcinoma, and importantly, there was no occurrence of lung metastasis (29). Furthermore, conditional loss of function of both copies of Pten in the thyroid results mainly in follicular adenoma within 12 months of age, which only progresses to invasive FTC at advanced ages, suggesting that even in a setting of maximal activation of PI3K-AKT signaling, other events are needed to trigger FTC (17). In contrast, the Pten+/−Inpp4b−/− mice developed aggressive, often metastatic, and always lethal follicular-like carcinoma by 5-6 months of age. The shorter latency to a more aggressive follicular-like carcinoma in Pten+/−Inpp4b−/− mice further support the notion that while increased Akt activation does play a role in follicular-like thyroid tumorigenesis, it is insufficient in mediating progression and metastases, and that the tumor suppressive function of INPP4B therefore extends beyond its role in suppressing the overall level of PI3K-AKT pathway activation. In this respect, the striking difference between PTEN and INPP4B that emerges from our study is their differential localization at the early endosome where INPP4B but not PTEN could regulate signaling in a localized and specialized fashion.
Beyond the level of AKT activation, isoform-specific AKT signaling plays an important role in mediating cancer progression. In the context of breast cancer, AKT2 has been implicated in metastasis, whereas AKT1 suppresses metastatic dissemination (30). Interestingly, signaling through AKT2 is critical for the development of thyroid neoplasms in Pten+/− mice (18). Specifically, loss of Akt2 in Pten+/− mice rescues the development of thyroid adenomas in this model (18). Furthermore, genomic amplification of AKT2 is frequently observed in FTC, but not in anaplastic thyroid carcinoma (31). These findings indicate that AKT2 activation is particularly important in follicular thyroid carcinoma progression and metastasis. Our finding that INPP4B, but not PTEN, localizes to the early endosomes to selectively regulate AKT2 identifies a novel and specific role for INPP4B in the regulation of PI3K-AKT signaling. INPP4B loss could therefore increase the PI(3,4)P2 pool in the early endosomes, regulate endocytic trafficking and contribute to prolonged AKT2 signaling from the endocytic membranes, through which it mediates its effects on thyroid carcinoma initiation, migration and invasion. Our findings are also in line with recent observation that activation of the endocytic trafficking pathways is critical for tumor cell migration (32). Therefore, it is possible that the aberrant activation of AKT2 at endosomes might represent the molecular mechanism underlying the characteristic metastatic propensity of FTCs. Intriguingly, although AKT1 is also localized at the early endosomes, INPP4B does not appear to regulate its activation. The exact mechanism underlying the selective regulation of AKT2 by INPP4B remains to be elucidated. Our study nevertheless represents an important first step in the understanding of the mechanism underlying isoform specific and localized AKT regulation.
In conclusion, our study provides strong evidence that INPP4B is not epistatic to PTEN and that INPP4B loss, although insufficient to initiate cancer in the thyroid, can promote FTC progression and metastasis in the context of PTEN haploinsufficiency through the isoform-specific regulation of AKT signaling at the endosomes (Fig. 6). More generally, our findings provide compelling evidence for the critical role of a qualitative regulation of signal transduction in tumorigenesis.
Figure 6. Model for the role of INPP4B and PTEN loss in FTC progression and metastasis.

INPP4B and PTEN control both activation level and subcellular localization of PI3K/AKT signalling to oppose thyroid tumorigenesis.
Methods
Mice and histopathological analyses
Inpp4b knockout mice were generated by Sasaki and colleagues. Total body necropsy and histopathological analyses were performed on cohorts of male and female mice from 4-15 months of age. Mouse tissues were fixed in 4% paraformaldehyde and embedded in paraffin. They were then sectioned and stained with hematoxylin and eosin (H&E) for pathological analyses. The use of these mice and procedures performed were in accordance to NIH-approved guideline, and the Institutional Animal Care and Use Committee (IACUC) ate Beth Israel Deaconess Medical Center approved the studies.
Studies with Primary cells
Mouse embryonic fibroblasts (MEFs) were isolated at day E13.5, immortalized with SV40 large T antigen, and maintained in DMEM supplemented with 10% fetal bovine serum, 2mM glutamine, 100 U/ml penicillin and streptomycin (Invitrogen).
Cell lines
All cells were maintained in DMEM or RPMI supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and streptomycin (Invitrogen). N-Thy-Ori-3 cells were purchased from Sigma. Htori and 8505C cells were kindly provided by Dr. Sareh Parangi (MGH); FTC and TPC1 cells were kindly provided by Dr. Orlo H. Clark (UCSF). Cell lines were tested for specific markers by Western blot and qRT-PCR in our laboratory, routinely tested for Mycoplasma (MycoAlert; Lonza), but not further authenticated.
Human tissue collection
The Committee for Human Research at Brigham and Women's Hospital approved this study. Thyroid tumors and normal tissue were discarded specimens obtained from patients undergoing thyroidectomy. The specimens were snap-frozen in the operating room suite with liquid nitrogen and were maintained at -80C until analysis. An endocrine pathologist, who confirmed the histologic diagnosis, evaluated the specimens.
Western blotting and immunohistochemistry
Cells and tissues were lysed with RIPA buffer (50mM Tris [pH8], 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40, 1mM EDTA and protease and phosphatase inhibitor cocktail [Roche]). For western blotting, the following antibodies were used: anti-AKT (9272, 1:1000), anti-p-AKT (pSer473, 9271, 1:1000; pThr308, 9275, 1:1000), anti-AKT1(2938, 1:1000), anti-AKT2(3063, 1:1000 and 5239, 1:1000), anti-p-AKT1 (pSer473)(9018, 1:1000), anti-p-AKT2 (pSer474)(8599, 1:1000), anti-PTEN (9559, 1:1000), anti-INPP4B(8450, 1:1000) were from Cell Signaling Technology. Please note that anti-AKT2 (3063) and (5239) cross-reacted with different unspecific bands, with a lower AKT2 specific band for (3063) and an upper specific band for (5239) as indicated. The specific bands are those that identified a protein of the expected molecular weight, while unspecific bands were migrating at very different molecular weights. Different and well-established antibodies against AKT2 (33, 34) were used in two independent laboratories in order to corroborate the reported findings. Anti-INPP4B (81269, 1:1000) was from Epitomics. Anti-HSP90 (610419, 1:3000) was from BD Biosciences anti-β-actin (A3853, 1:5000) was from Sigma. The characterization of anti-INPP4B (81269) has been shown in the Suppl Fig. 2. p-AKT1 and p-AKT2 band intensities were normalized to the corresponding AKT1 and AKT2 expression levels, except in Figure 4G where they were normalized to RAB5. Band intensities were quantified with ImageJ software. For immunohistochemistry, anti-thyroglobulin antibody (80783, 1:2000) was from Abcam and anti-p-Akt (pSer473, 4060, 1:100) was from Cell Signaling Technology.
Immunofluorescence
Cells were grown on coverslips, fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton or pre-chilled methanol. Cells were rinsed with PBS, blocked and then incubated with primary antibody, followed by incubation with Alexa Fluor conjugated secondary antibodies (Life Techonologies). Coverslips were mounted with ProLong Gold Antifade reagent with DAPI (Life Technologies). Confocal images of cells were acquired with LSM510META Confocal Laser System (BIDMC) and Zeiss Observer-Z1 microscope equipped with Apotome (University of Torino). Primary antibodies used: anti-INPP4B (Atlas, 37682, 1:50), anti-PI3K-C2α (anti-p170) (BD, 611046, 1:25) anti-γ-tubulin (Sigma, GTU-88, 1:100), anti-Rab5 (BD, 610261, 1:25 and Cell Signaling Technology, 3547, 1:50) anti-α-tubulin (Sigma, T6074, 1:100), anti-PtdIns(3,4P)2 and anti-PtdIns(3,4,5)P3 (Echelon, Z-P034b, 1:50 and Z-P345, 1:50), anti-SNX9 (Proteintech, 15721-1-AP, 1:400), and anti-AKT2 (Cell Signaling Technology, 3063, 1:100).
Early endosome purification
Cells were gently homogenized in the homogenization buffer (250mM sucrose, 3mM imidazole, pH 7.4 with protease inhibitor cocktail). The samples were centrifuged at 3000 rpm to remove nuclei and cell debris. Postnucelar supernatant (PNS) was subsequently separated by sucrose gradient centrifugation into different cellular fractions. In detail, the PNSs were adjusted to 40.6% sucrose using a stock solution (62% sucrose, 3mM imidazole pH 7.4), loaded at the bottom of centrifugation tubes (SW55), then sequentially overlaid with 1.5 ml 35% sucrose solution (35% sucrose, 3mM imidazole pH 7.4) followed by 1ml 25% solution (25% sucrose, 3mM imidazole pH 7.4) and 1ml of homogenization buffer on top of the load. After 1hour centrifugation, at 35000 rpm 4°C, early endosomes (EE) were recovered from interphase between 35% and 25% layers, late endosomes (LE) were recovered from uppermost portion of 25% phase, and heavy membranes (HM) including ER, Golgi and plasma membranes were recovered from lowest interphase. EE, LE and HM were then precipitated with methanol/chloroform loaded in SDS-PAGE for western blot analyses.
RNA isolation and RT-qPCR
Total RNA was purified from cell lines and tissues using the PureLink RNA Mini Kit (Invitrogen). For qPCR analysis, 2ug of total RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). SYBR-Green qPCR analysis was then performed using Applied Biosystems StepOnePlus in accordance to the manufacturer's protocol. Each target was run in triplicate, and expression levels were normalized to mouse hypoxanthine-guanine phosphoribosyltransferase (HPRT) or human porphobilinogen (PBGD).
Genotyping
The following genotyping primers were used:
Inpp4b del1: GTTTACATTTGACAGGGTGGTTGG
Inpp4b del2: TGCTGTCGCCGAAGAAGTTA
Inpp4b del3: CCTGCCATGGGTAGATTTCT
Pgen-1: TGGGAAGAACCTAGCTTGGAGG
Pgen-3: ACTCTACCAGCCCAAGGCCCGG
3193: CGAGACTAGTGAGACGTGCTACTTCC
5-Aza-2′-deoxycytidine treatment
Cells were briefly treated with 3uM of 5-Aza-2′-deoxycytidine for 5 days. After that, the cells were harvested for RNA and protein analysis.
Growth proliferation assay
Cells were plated at a density of 2.5×104 cells/well in 12-well plates and each sample was plated in triplicate. Plates were collected on day 0, day 2, day 4 and day 6. The wells were washed with PBS and cells were fixed with 4% paraformaldehyde (Santa Cruz Biotechnology). Wells were stained with 0.1% Crystal Violet solution, washed and dried. The absorbed stained was then solubilized with 10% acetic acid, and the absorbance was measured at 595nm.
Soft-agar colony formation assay
Soft-agar colony formation assay was performed by first plating 6-well tissue culture plates with 0.6% Noble agar/growth media and allowed to solidify at room temperature. 1×105 thyroid cancer cell lines in 0.3% Noble agar/growth media were then seeded as the top layer. Each cell line was seeded in triplicate. The soft agar was allowed to solidify at room temperature, then placed in the incubator at 37°C. Fresh growth media was added every week, and colonies were counted and photographed after 2 weeks.
Measurement of TSH levels
Serum was collected from, Pten+/−, Pten+/−Inpp4b+/− and Pten+/−Inpp4b−/− mice. The mice were between 3-5 months of age, and at least 4 mice in each genotype were tested. Briefly, blood was allowed to clot at 4°C for at least 2 hours. It was then centrifuged at 1000×g for 15 minutes. The serum was carefully removed and frozen at -20°C. For testing, we used the ultrasensitive thyroid-stimulating hormone (U-TSH) ELISA kit from MyBioSource (MBS042764).
Bisulfite sequencing
Genomic DNA samples were collected and treated with bisulfite using the EpiTect Bisulfite kit (Qiagen) according to the manufacturer's recommendations. PCR amplification was performed with primers specific for the methylated and unmethylated alleles, as described in Yuen et al. (12).
Statistical analysis
For quantitative data, data sets were generally analyzed using the unpaired, two-tailed Student's t tests (GraphPad Prism, GraphPad Software). p<0.05 was considered significant.
Supplementary Material
Significance.
Although both PTEN and INPP4B can inhibit PI3K/AKT signaling through their lipid phosphatase activities, here we demonstrate lack of an epistatic relationship between the two tumor suppressors. Instead, the qualitative regulation of PI3K/AKT2 signaling by INPP4B provides a mechanism for their cooperation in suppressing thyroid tumorigenesis and metastasis.
Acknowledgments
The authors thank all members of the Pandolfi laboratory for critical discussion, Lauren Fawls for editing the manuscript, Kelsey Berry for technical assistance and Justine Barletta for help with histopathological interpretation. The authors are grateful to Min Sup Song and Su Jung Song for insightful discussion.
Grant Support: This work has been supported by NIH grant U01 CA141496 to P.P.P. C.L.C. was supported by the A*STAR National Science Scholarship (Singapore).
Footnotes
Authors' Contributions: Conception and design: C.L. Chew, D.T. Ruan, A. Lunardi, L. Salmena, P.P. Pandolfi
Development of methodology: C.L. Chew, F. Gulluni, D.T. Ruan, A. Lunardi, M. Chen, L. Salmena, A. Papa
Acquisition of data: C.L. Chew, F. Gulluni, D.T. Ruan, M. Chen, C. Ng, J. Fung
Analysis and interpretation of data (e.g. statistical analysis, H&E and IHC interpretation): C.L. Chew, D.T. Ruan, A. Lunardi, M. Chen, M. Nishino, R.T. Bronson, E. Hirsch, P.P. Pandolfi
Writing, review, and/or revision of the manuscript: C.L. Chew, D.T. Ruan, A. Lunardi, M. Chen, P.P. Pandolfi
Administrative, technical, or material support: C.L. Chew, D.T. Ruan, A. Lunardi, A. Papa, J.G. Clohessy, J. Sasaki, T. Sasaki, L. Longo
Study supervision: C.L. Chew, A. Lunardi, D.T. Ruan, A. Papa, P.P. Pandolfi
Carried out experiments: C.L. Chew, F. Gulluni, M. Chen, A. Lunardi
Provided histology and immunohistochemistry: C. Ng, J. Fung
Conflict of Interest: None of the authors have anything to declare
References
- 1.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews Drug discovery. 2014;13:140–56. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Advances in biological regulation. 2014;55:28–38. doi: 10.1016/j.jbior.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, et al. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer research. 2003;63:196–206. [PubMed] [Google Scholar]
- 4.Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer research. 2004;64:3171–8. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 5.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. The Journal of cell biology. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nature genetics. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 7.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nature reviews Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–48. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 9.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer cell. 2009;16:115–25. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fedele CG, Ooms LM, Ho M, Vieusseux J, O'Toole SA, Millar EK, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:22231–6. doi: 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Lorenzo R, Gill KZ, Shen CH, Zhao FX, Zheng B, Schulze HJ, et al. A tumor suppressor function for the lipid phosphatase INPP4B in melanocytic neoplasms. The Journal of investigative dermatology. 2014;134:1359–68. doi: 10.1038/jid.2013.511. [DOI] [PubMed] [Google Scholar]
- 12.Yuen JW, Chung GT, Lun SW, Cheung CC, To KF, Lo KW. Epigenetic inactivation of inositol polyphosphate 4-phosphatase B (INPP4B), a regulator of PI3K/AKT signaling pathway in EBV-associated nasopharyngeal carcinoma. PloS one. 2014;9:e105163. doi: 10.1371/journal.pone.0105163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodgson MC, Shao LJ, Frolov A, Li R, Peterson LE, Ayala G, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer research. 2011;71:572–82. doi: 10.1158/0008-5472.CAN-10-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science (New York, NY) 1997;275:665–8. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 16.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/- mice. Science (New York, NY) 1999;285:2122–5. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 17.Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene. 2010;29:5678–86. doi: 10.1038/onc.2010.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu PZ, Chen ML, Jeon SM, Peng XD, Hay N. The effect Akt2 deletion on tumor development in Pten(+/-) mice. Oncogene. 2012;31:518–26. doi: 10.1038/onc.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walz HA, Shi X, Chouinard M, Bue CA, Navaroli DM, Hayakawa A, et al. Isoform-specific regulation of Akt signaling by the endosomal protein WDFY2. The Journal of biological chemistry. 2010;285:14101–8. doi: 10.1074/jbc.M110.110536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franco I, Gulluni F, Campa CC, Costa C, Margaria JP, Ciraolo E, et al. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Developmental cell. 2014;28:647–58. doi: 10.1016/j.devcel.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature. 2013;499:233–7. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 22.Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer research. 2001;61:6105–11. [PubMed] [Google Scholar]
- 23.Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. The Journal of clinical endocrinology and metabolism. 2011;96:E2063–71. doi: 10.1210/jc.2011-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furuya F, Hanover JA, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1780–5. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pringle DR, Vasko VV, Yu L, Manchanda PK, Lee AA, Zhang X, et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. The Journal of clinical endocrinology and metabolism. 2014;99:E804–12. doi: 10.1210/jc.2013-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, et al. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology. 2005;146:4456–63. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 27.Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, 2nd, Tallini G, et al. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. The Journal of clinical endocrinology and metabolism. 2003;88:2318–26. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- 28.Diallo-Krou E, Yu J, Colby LA, Inoki K, Wilkinson JE, Thomas DG, et al. Paired box gene 8-peroxisome proliferator-activated receptor-gamma fusion protein and loss of phosphatase and tensin homolog synergistically cause thyroid hyperplasia in transgenic mice. Endocrinology. 2009;150:5181–90. doi: 10.1210/en.2009-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157:595–610. doi: 10.1016/j.cell.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer research. 2009;69:5057–64. doi: 10.1158/0008-5472.CAN-08-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. The Journal of clinical endocrinology and metabolism. 2008;93:3106–16. doi: 10.1210/jc.2008-0273. [DOI] [PubMed] [Google Scholar]
- 32.Palamidessi A, Frittoli E, Garre M, Faretta M, Mione M, Testa I, et al. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell. 2008;134:135–47. doi: 10.1016/j.cell.2008.05.034. [DOI] [PubMed] [Google Scholar]
- 33.Chin RY, Yuan X, Balk SP, Toker A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer discovery. 2014;4:942–55. doi: 10.1158/2159-8290.CD-13-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ullal AV, Peterson V, Agasti SS, Tuang S, Juric D, Castro CM, et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Sci Transl Med. 2014;6:219ra9. doi: 10.1126/scitranslmed.3007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.