

Abstract
Background
A highly pathogenic human coronavirus (CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), has emerged in Jeddah and other places in Saudi Arabia, and has quickly spread to European and Asian countries since September 2012. Up to the 1st October 2015 it has infected at least 1593 people with a global fatality rate of about 35%. Studies to understand the virus are necessary and urgent. In the present study, MERS-CoV main protease (Mpro) is expressed; the dimerization of the protein and its relationship to catalysis are investigated.
Methods and Results
The crystal structure of MERS-CoV Mpro indicates that it shares a similar scaffold to that of other coronaviral Mpro and consists of chymotrypsin-like domains I and II and a helical domain III of five helices. Analytical ultracentrifugation analysis demonstrated that MERS-CoV Mpro undergoes a monomer to dimer conversion in the presence of a peptide substrate. Glu169 is a key residue and plays a dual role in both dimerization and catalysis. The mutagenesis of other residues found on the dimerization interface indicate that dimerization of MERS-CoV Mpro is required for its catalytic activity. One mutation, M298R, resulted in a stable dimer with a higher level of proteolytic activity than the wild-type enzyme.
Conclusions
MERS-CoV Mpro shows substrate-induced dimerization and potent proteolytic activity. A critical assessment of the residues important to these processes provides insights into the correlation between dimerization and catalysis within the coronaviral Mpro family.
Introduction
A highly pathogenic human coronavirus (CoV)1, Middle East respiratory syndrome coronavirus (MERS-CoV), emerged in Jeddah and other places in Saudi Arabia in September 2012 and spread to some European, African and Asian countries in recent years [1–3]. The virus causes symptoms similar to severe acute respiratory syndrome coronavirus (SARS-CoV), but an additional component is also involved, namely acute renal failure [4]. Up to the 1st October 2015, 1593 people had been infected with MERS and this has led to 568 reported deaths globally (World Health Organization, global alert and response, http://www.who.int/csr/don/01-october-2015-mers-jordan/en/). A serological study of major livestock in Saudi Arabia suggested dromedary camels may be an original reservoir [5, 6]; although recent studies have identified bats may also as the suspected host as a number of bat CoVs show high sequence similarity to SARS-CoV and MERS-CoV [7, 8] and a bat-CoV was discovered that readily infect human cells using ACE2 as the receptor [9]. Nevertheless, human-to-human transmission of MERS-CoV has been confirmed. This May, an infected traveler from the Middle East region returned to his home country of the Republic of Korea and caused an outbreak that revolved around health facilities (http://www.who.int/csr/don/07-july-2015-mers-korea/en/). These findings indicate that it is possible for the virus to spread globally and as such it poses a significant threat to world health and the world economy in general. Therefore studies that help to understand this virus and aid the development of antiviral drugs or other therapies are important.
Similar to other CoVs, after entering the host cells, the nonstructural polyproteins (pp1a and pp1ab) of MERS-CoV are synthesized and then cleaved by two coronaviral proteases, a main protease (Mpro) (EC 3.4.22.69) and a papain-like protease (EC 3.4.22.46) [10]. This cleavage is considered to be a leading process which is required for viral maturation [11–14]. The MERS-CoV Mpro, namely nsp5 of the pp1a proteins (residue 3248–3553), has been identified [15]. Like other Mpro, there is a catalytic dyad that consists of a His residue and a Cys residue [16–20]. Sequence alignment suggests that MERS-CoV Mpro, in a similar manner to other known Mpro, has a chymotrypsin-like architecture consisting of a catalytic core (domain I and II) and a helical domain III; sequence identities of CoV Mpro protein range from 50% to 80% (S1 Fig). Recently the crystal structure of an inactive MERS-CoV Mpro C148A mutant has confirmed this similarity and the results also suggest that this protein forms a dimer [15]. Furthermore, based on the identification of eleven canonical cleavage sites, the MERS-CoV Mpro should be able to recognize and cleave at the (L/M)-Q-↓-(A/S) conserved sequence, which is essential for most CoV Mpro-mediated processing [21, 22]. However, up to now, the correlation between the protein’s structure and the catalytic process remains unclear.
In the present study we expressed and purified the MERS-CoV Mpro using the authentic N-terminus via an Escherichia coli system. The crystal structure of the MERS-CoV Mpro at 3.0-Å resolution is reported. The quaternary structural changes in the MERS-CoV Mpro in the absence and presence of peptide substrates were investigated by analytical ultracentrifugation (AUC). The results of kinetic activity assays indicated that MERS-CoV Mpro exhibits potent proteolytic activity that is associated with a pattern of cooperativity. Some critical residues for dimerization of the protein and catalysis by the protein were verified by site-directed mutagenesis. The present studies provide a foundation for an understanding of the mechanism that controls the monomer-dimer switch at work in MERS-CoV Mpro.
Materials and Methods
Expression Plasmid Construction
The sequence of the MERS-CoV Mpro (GenBank accession number AHC74086; polyprotein residues 3248–3553) was synthesized (MDBio Inc.), digested by NdeI-XhoI and then inserted into the vector pET-28a(+) (Novagen). In this construct, the 6 x His tag is retained at the N-terminus. To remove the fusion tag and generate an authentic N-terminus for protein purification, the codons of the thrombin cutting recognition sequence and a NdeI cutting site were removed and then inserted the codons of Leu-Arg-Leu-Lys-Gly-Gly into the above vector. The forward primer sequence for site-directed mutagenesis was 5’-CATCACAGCAGCGGCCTGCGTCTGAAAGGCGGCAGCGGTTTGGTGAAAATG-3’ and the reverse primer was 5’-CATTTTCACCAAACCGCTGCCGCCTTTC AGACGCAGGCCGCTGCTGTGATG-3’. The reading frame of the final plasmid was confirmed by sequencing.
Expression and Purification of MERS-CoV Mpro
The expression vector was transformed into E. coli BL21 (DE3) cells (Novagen). Cultures were grown in 0.8 liters of LB medium at 37°C for 4 h, induced with 0.4 mM isopropyl-β-D-thiogalactopyranoside, and then incubated overnight at 20°C. After centrifuging at 6,000 x g at 4°C for 15 min, the cell pellets were resuspended in lysis buffer (20 mM Tris, pH 8.5, 250 mM NaCl, 5% glycerol, 0.2% Triton X-100, and 2 mM β-mercaptoethanol) and then lysed by sonication. The crude extract was then centrifuged at 12,000 x g at 4°C for 25 min to remove the insoluble pellet. Next the supernatant was incubated with 1-ml Ni-NTA beads at 4°C for 1 h and then loaded onto an empty column. After allowing the supernatant to flow through, the beads were washed with washing buffer (20 mM Tris, pH 8.5, 250 mM NaCl, 8 mM imidazole, and 2 mM β-mercaptoethanol). The SARS-CoV papain-like protease [12] (1 mg in 100 mM phosphate buffer (pH 6.5)) was then added and incubated for 3 h. The SARS-CoV papain-like protease digestion, which removed the 6 x His tag and Leu-Arg-Leu-Lys-Gly-Gly fragment, resulted in a native protein product with an authentic N-terminus. The digest was allowed to flow through and then loaded onto a S-100 gel-filtration column (GE Healthcare) equilibrated with running buffer (20 mM Tris, pH 8.5, 100 mM NaCl, and 2 mM dithiothreitol). The purity of the fractions collected was analyzed by SDS-PAGE and the protein was concentrated to 30 mg/ml by Amicon Ultra-4 10-kDa centrifugal filter (Millipore).
Protein Crystallography
Crystals of the MERS-CoV Mpro were obtained at 295 K by the sitting-drop vapor-diffusion method. The protein solution was set up at 5 mg/ml and the reservoir solution consisted of 0.1 M Tris, pH 8.4, 15% (w/v) PEG 4000 and 0.2 M sodium acetate. Clusters of needle crystals appeared in 2 days and were used for micro-seeding. Single cystals of rectangle shape and with dimensions of 0.3–0.5 mm were obtained in less than a week. All crystals were cryoprotected in the reservoir solution with 15% glycerol and were flash-cooled in liquid nitrogen.
Data collection, structure determination and refinement
X-ray diffraction data were collected at 100 K on the SPXF beamline 13C1 at the National Synchrotron Radiation Research Center, Taiwan, ROC, using a ADSC Quantum-315r CCD detector (X-ray wavelength of 0.976 Å). The diffraction images were processed and scaled using the HKL-2000 package [23]. The structure was solved by the molecular replacement method by Phaser [24] using the structure of SARS-CoV Mpro R298A mutant (PDB entry 4hi3; [25]) as the search model. Manual rebuilding of the structure model was performed using Coot [26]. Structure refinement was carried out using REFMAC [27]. The data-processing and refinement has been deposited in the Protein Data Bank (PDB entry 5c3n).
Steady-State Kinetic Analysis
The colorimetry-based peptide substrate, TSAVLQ-para-nitroanilide (TQ6-pNA) (purity 95–99% by HPLC; GL Biochem Ltd, Shanghai, China), was used to measure the proteolytic activity of MERS-CoV Mpro and its mutants throughout the course of the study as described previously [25, 28]. This substrate is cleaved at the Gln-pNA bond to release free pNA, resulting in an increase in absorbance at 405 nm. The absorbance at 405 nm was continuously monitored using a Jasco V-550 UV/VIS spectrophotometer. The protease activity assay was performed in 10 mM phosphate (pH 7.6) at 30°C. The substrate stock solution was 1600 μM and the working concentrations were from 25 to 1200 μM. In the substrate titration assay, the concentration of MERS-CoV Mpro and its mutants, V4R, T126S, E169A, M298R and T126S/M298R was 0.3, 0.4, 0.7, 1.2, 0.15 and 0.26 μM, respectively, while that of SARS-CoV Mpro was 1.1 μM. Steady state enzyme kinetic parameters were obtained by fitting the initial velocity (ν0) data to the Michaelis-Menten Eq (1)
| (1) |
where kcat is the catalytic constant, [E] is the enzyme concentration, [S] is the substrate concentration and Km is the Michaelis constant of the substrate. The program SigmaPlot (Systat Software, Inc., Richmond, CA) was used for the data analysis.
To assess the cooperativity effect, the kinetic parameters were obtained by fitting the initial velocities to the Hill Eq (2)
| (2) |
where K’ is a constant that is related to the dissociation constant and h is the Hill constant.
Analytical ultracentrifugation analysis
AUC was performed on a XL-A analytical ultracentrifuge (Beckman Coulter) using an An-50 Ti rotor [11, 12, 25, 28–30]. The sedimentation velocity experiments were carried out using a double-sector epon charcoal-filled centerpiece at 20°C with a rotor speed of 42,000 rpm. Protein solutions of 0.05 to 0.5 mg/ml (330 μl) and reference (370 μl) solutions, both containing D2O, were loaded into the centerpiece. The absorbance at 280 nm was monitored in a continuous mode with a time interval of 300 s and a step size of 0.003 cm. Multiple scans at different time intervals were then fitted to a continuous c(s) distribution model using the SEDFIT program [31]. Additionally, the results with the various different protein concentrations were globally fitted to a monomer-dimer self-association model using the SEDPHAT program to calculate the dissociation constant (Kd) [32].
To measure the substrate-induced dimerization, the active enzyme centrifugation (AEC) [33] was performed. Briefly, MERS-CoV Mpro of 15 μl (1 mg/ml) was added into the small well of the band-forming centerpiece before the cell assembled. Then 330 μl of peptide substrate at 0, 200 and 400 μM in D2O were respectively loaded into the bulk sample sector space. At a rotor speed of 42,000 rpm, the protein solution flowed into the substrate-containing channel and form a protein band. It can be detected by absorbance at 250 nm. During the centrifugation, the sediment protein continuously met and cleaved the substrate, which can be detected by absorbance change at 405 nm. The dataset from the multiple scans at 250 nm at various time intervals were fitted to a continuous c(s) distribution model using the SEDFIT program [31], while the first five scans (0–30 min) at 405 nm were used to derive the product concentration and then initial velocity values.
Isothermal titration calorimetry (ITC)
The protocol followed that of previous studies [28] with some modifications. Apparent dissociation constants and stoichiometry of the enzyme-ligand interactions were measured by a Thermal Activity Monitor 2277 from TA instruments (New Castle, DE). Calorimetric titrations of the peptide substrate TQ6-pNA (0.5 mM in a 250-μl syringe) and Mpro (6 μM in a 4-ml ampoule) were carried out at 25°C in 10 mM phosphate buffer (pH 7.6). The peptides were titrated into the enzyme using a 10-μl aliquot for each injection with a time interval of 20 min. A control experiment in the absence of enzyme was performed in parallel to correct for the dilution of heat. The data obtained was then analyzed by integrating the heat effects normalized against the amount of injected protein using curve-fitting based on a 1:1 binding model. This involved the use of Digitam software (TA instruments, New Castle, DE).
Results and Discussion
Recombinant MERS-CoV Mpro preparation
As part of the present study, an expression vector was constructed and the BL21 (DE3) STAR (Invitrogen) strain of E. coli were used to express MERS-CoV Mpro. Unlike SARS-CoV Mpro [25, 28], the MERS-CoV Mpro with 6 x His-tag retained at the C-terminus cannot be expressed. Instead, the bacteria are able to express the Mpro when there is a N-terminal 6 x His-tag fusion that can be removed during the purification. However, thrombin digestion leaves two extra residues (Gly-Ser) at the N-terminus of Mpro, resulting in protein with no proteolytic activity (data not shown). Therefore we used SARS-CoV papain-like protease [12, 30, 34], which is a highly active viral deubiquitinase and does not leave any residues at the N-terminus of Mpro. After gel-filtration, the purity of authentic N-terminus Mpro was about 99% (S2 Fig). The size of the MERS-CoV Mpro was found to be close to 30 kDa, while any uncut protein was located at higher molecular weight position. The typical yield was about 10 mg after purification from 0.8 liter of E. coli culture.
Overall structure of MERS-CoV Mpro
The structure of the MERS-CoV Mpro was determined at 3.0 Å resolution by X-ray crystallography (Table 1 and Fig 1A). The crystal packing belonged to space group C2221, with unit-cell parameters a = 87.2, b = 94.0, c = 155.1 Å and α = β = γ = 90°. The final atomic model containing two Mpro molecules in a crystallographic asymmetric unit agrees well with the crystallographic data and the expected values of geometric parameters (Table 1). There are no residues in the disallowed region of the Ramachandran plot, while 81.3% of the residues are in the most favored region.
Table 1. Summary of crystallographic information for MERS-CoV Mpro.
| Data Collection | |
|---|---|
| Space group | C2221 |
| Cell dimensions | |
| a, b, c (Å) | 87.2, 94.0, 155.1 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution a (Å) | 30–3.0 (3.11–3.0) |
| R merge b (%) | 17 (62.9) |
| I / σI | 10.2 (3.1) |
| Completeness (%) | 94.6 (95.2) |
| Redundancy | 5.5 (5.6) |
| Refinement | |
| Number of reflections | 11,675 (1,549) |
| R factor c (%) | 21.8 |
| Free R factor d (%) | 28.2 |
| Number of atoms | |
| Protein | 4,580 |
| Water | 0 |
| Average B-factors for protein atoms (Å2) | 81.6 |
| R.m.s. deviations | |
| Bond length (Å) | 0.009 |
| Bond angles (°) | 1.5 |
| Ramachandran plot statistics (%) | |
| Most favored region | 81.3 |
| Additional allowed region | 13.6 |
| Generously allowed region | 5.2 |
| Disallowed region | 0 |
a The numbers in parentheses are for the highest-resolution shell.
b , where I hi is the integrated intensity of a given reflection and 〈I h〉 is the mean intensity of multiple corresponding symmetry-related reflections.
c , where and are the observed and calculated structure factors, respectively.
d Free R is R calculated using a random 5% of data excluded from the refinement.
Fig 1. The structure of MERS-CoV Mpro.

(A) The overall structure of the dimeric Mpro. The two protomers are shown as a ribbon and as a surface model, respectively. The negatively and positively charged regions on the molecular surfaces are colored red and blue. The spheres indicate the catalytic dyad, His41-Cys148. (B) Stereo view of an overlay of the dimerization interface and the active site of MERS-CoV Mpro (in cyan and orange) with that of SARS-CoV Mpro (grey; [16]). The red dashed lines indicate polar interactions between the two protomers of MERS-CoV Mpro, while the black dashed lines show polar interactions between those of SARS-CoV Mpro. The C atoms of the modeled substrate P4-P1 residues (from the structure of C148A mutant [15]) are colored magenta. The structural figures in this paper were produced using PyMol (http://www.pymol.org/).
The overall dimeric structure of Mpro is similar to that of SARS-CoV Mpro; although a relative shift of 10° to 30° could be observed for the two domain III within the dimer (S3A Fig). Indeed, the r.m.s. distance between equivalent Cα atoms of the domain I+II of the two structures is 0.9 Å, while that between the domain III of the two structures is 3.1 Å. Compared with the structures of the ligand-bound complex (PDB entry 4YLU) and C148A mutant (PDB entry 4WME) [14, 15], the r. m. s. distance is 0.8 and 0.7 Å over 540 Cα atom pairs, respectively (S3B Fig). This indicates that the dimeric structures show no significant difference; although the present structure is a free enzyme and the other two structures involve enzyme-ligand complexes and higher resolution. Besides, there is minor difference between the present structure and bat-CoV HKU4 Mpro (PDB entry 2YNA), with 80% sequence identity, as the r. m. s. distance over 539 Cα atom pairs is 0.8 Å (S3B Fig).
Interestingly, the dimerization interface situation with the MERS-CoV Mpro was found to be different to that of SARS-CoV Mpro where there are four amino acid pairs with intermolecular polar interactions (Ser1…Glu166, Arg4…Glu290, Ser123…Arg298 and Ser139…Gln299). There are only two pairs of intermolecular hydrogen bonds, Ser1…Glu169 and Ser142…Gln299 that are associated with the dimer surface of MERS-CoV Mpro according to the current structure (Fig 1B). This led us to compare the dimerization and catalytic activity of the two types of Mpro. In the present study, in addition to using the wild-type MERS-CoV Mpro, we also mutated several residues at the dimerization interface in order to evaluate their role in dimerization and catalysis of MERS-CoV Mpro (see below).
Correlation between dimerization and catalysis of MERS-CoV Mpro
To compare catalysis between the two Mpro, TQ6-pNA, a peptide substrate for SARS-CoV Mpro [25, 28], was used to measure the proteolytic activity. At first, the dependence of the initial velocity on enzyme concentration was analyzed and showed a nonlinear upward correlation (Fig 2A). The pattern is similar to that of SARS-CoV Mpro, as the monomeric Mpro may not have catalytic activity [28]. However, MERS-CoV Mpro displayed a sigmoid curve for its rate constant pattern at various substrate concentrations (Fig 2B, open circles); this contrast with SARS-CoV Mpro, which exhibited a classical saturation curve (S4 Fig). The results were then fitted to the Hill equation (Eq 2) in order to evaluate the kinetic parameters (Table 2). The kcat (2.33 s-1) of MERS-CoV Mpro is close to that of SARS-CoV Mpro (2.11 s-1), while the Hill constant was 1.8, suggesting a significant degree of positive cooperativity among the Mpro protomers. The comparable activity levels of the two Mpro in the present study is dissimilar to the results obtained during a recent study in which the activity of the MERS-CoV Mpro was found to be 5-fold lower than that of SARS-CoV [14]. Using different substrates may cause the difference. Tomar et al. [14] used a longer peptide substrate with residues present at both P and P’ site. However, in the present studies we used a peptide substrate that contains only P site residues. Besides, they utilized FRET substrate and could only be used at low substrate concentrations to prevent the inner-filter effect, while we are able to use higher substrate concentrations to capture the kinetic parameters.
Fig 2. Proteolytic activity assay of MERS-CoV Mpro and its mutants.

(A) Plot of initial velocities (vi) versus the concentration of wild-type MERS-CoV Mpro. The concentration of substrate (TQ6-pNA) was 600 μM. The line represented the best-fit results according to the nonlinear dependence equation [28]. (B) and (C) Plots of rate constant (kobs) versus the concentration of peptide substrate for MERS-CoV Mpro (by circles), the V4R mutant (by lower triangles), the T126S mutant (by squares), the E169A (by diamonds), the M298R (by upper triangles) and the T126S/M298R (by hexagons) are indicated. The lines represented the best-fit results according to the Hill equation (Eq 2; wild-type, V4R, T126S and E169A mutants) or the Michaelis-Menten equation (Eq 1; M298R and T126S/M298R mutants). The protein concentrations of the wild-type, V4R, T126S, E169A, M298R and T126S/M298R mutants used for the assay were 0.3, 0.4, 0.7, 1.2, 0.15 and 0.26 μM, respectively. All the assays were performed in 10 mM phosphate (pH7.6) at 30°C and repeated twice to ensure reproducibility and the error bars were shown. The kinetic parameters are shown in Table 2.
Table 2. The kinetic parameters and dissociation constants of MERS-CoV Mpro.
| Proteins | Kinetic Parameters a | Dissociation Constant b | |||
|---|---|---|---|---|---|
| Km (μM) | k cat (s-1) | h | No substrate(μM) | With 600 μM substrate (μM) | |
| MERS-CoV Mpro | |||||
| Wild-type | - | 2.33 ± 0.13 | 1.8 ± 0.04 | 7.7 ± 0.3 | 0.7 ± 0.04 |
| V4R | - | 0.96 ± 0.05 | 2.7 ± 0.2 | 23.0 ± 0.4 | 15.2 ± 0.3 |
| T126S | - | 0.56 ± 0.04 | 2.0 ± 0.2 | 33.7 ± 0.9 | 13.9 ± 0.1 |
| E169A | - | 0.41 ± 0.02 | 2.1 ± 0.1 | 14.3 ± 0.2 | 14.1 ± 0.5 |
| M298R | 181.0 ± 24.0 | 7.91 ± 0.49 | - | 1.1 ± 0.1 | 0.7 ± 0.01 |
| T126S/M298R | 419.4 ± 63.9 | 4.63 ± 0.37 | - | 2.8 ± 0.1 | 0.9 ± 0.01 |
| SARS-CoV Mpro | 890 ± 130 | 2.11 ± 0.15 | - | 0.7 ± 0.02 | 1.7 ± 0.03 c |
a Kinetic data of SARS-CoV Mpro and MERS-CoV M298R and T126S/M298R mutants were fitted to the Michaelis-Menten equation (Eq 1), while those of the others were fitted to the Hill equation (Eq 2). The Rsqr were from 0.985 to 0.999, respectively. All the assays were repeated twice and the average values were used for the fitting.
b The values were derived from a global fit of the AUC data to a monomer-dimer self-association model by SEDPHAT [32]. The experiments for the assay were obtained at protein concentration of 1.5 to 30 μM.
c The value was from our previous studies for comparison [28].
The cooperativity phenomenon associated with the MERS-CoV Mpro is similar to that of the SARS-CoV Mpro R298A/L monomer mutants; these were found to show monomer to dimer conversion during catalysis [28]. As a result of the above, we investigated the quaternary structure of the Mpro by AUC (Fig 3). The cumulative spectra (Fig 3A) were analyzed using the continuous c(s) distribution model and the results suggested that MERS-CoV Mpro is a monomer in phosphate buffer (S5 Fig) and this contrasts with a distribution of 30% monomer and 70% dimer in the presence of 600 μM TQ6-pNA (Fig 3B). We also measured the size distribution of Mpro at various TQ6-pNA concentrations (S6 Fig). The results indicated that the sedimentation coefficient of the major species was shifted as the substrate dosage changed (S6B Fig). More substrate led to the major species moving close to the dimer position.
Fig 3. The continuous size distribution change of MERS-CoV Mpro.
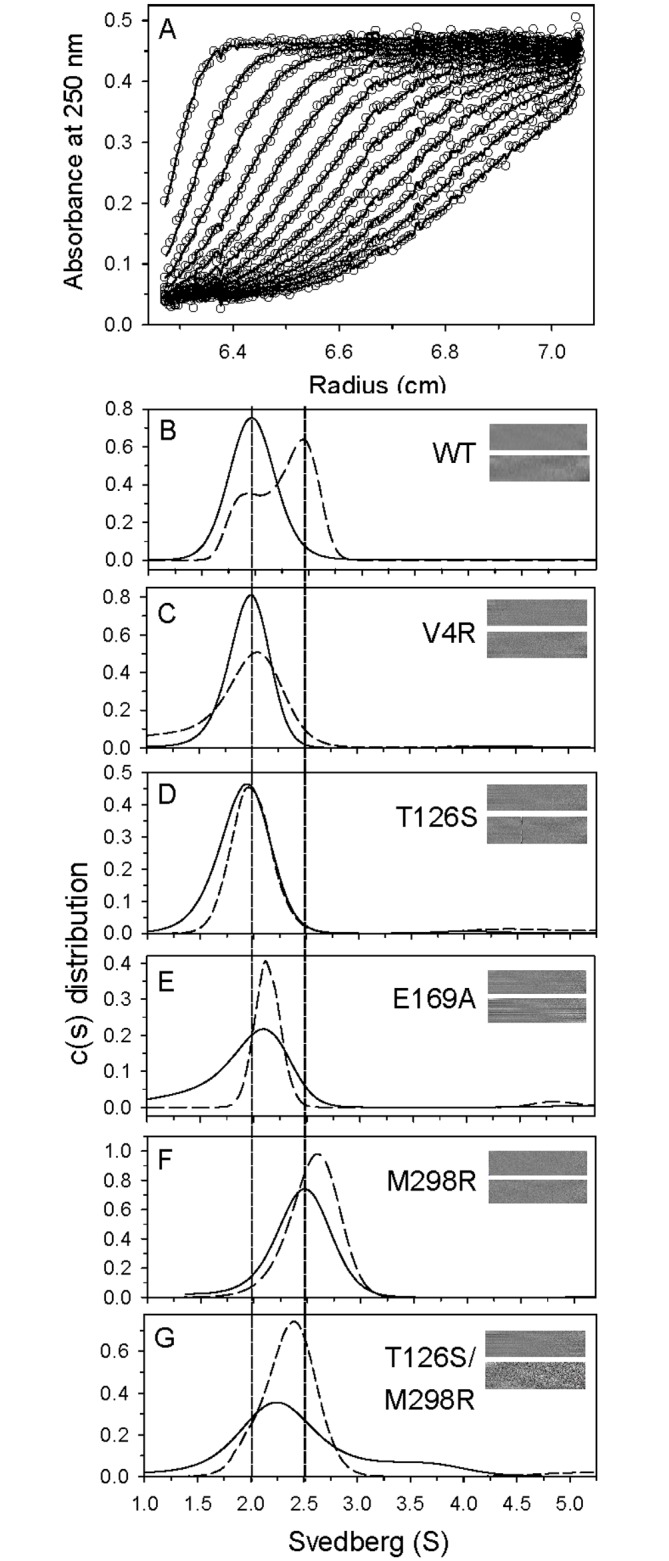
(A) Typical trace of absorbance at 250 nm of the enzyme during the SV experiment. The protein concentration was 0.5 mg/ml. For clarity, only every fifth scan is shown. The symbols represent the experimental data and the lines represent the results fitted to the Lamm equation using the SEDFIT program [31]. (B-G) Continuous c(s) distribution of wild-type, V4R, T126S, E169A, M298R and T126S/M298R mutants. The distributions in D2O containing 10 mM phosphate (pH 7.6) are shown by solid lines and those in the same buffer but with 600 μM TQ6-pNA substrate are shown by dashed lines. The left vertical dotted line indicates the monomer position and the right dotted line represents the dimer position. The residual bitmap of the raw data and the best-fit results are shown in the insets.
However, before the centrifugation, the enzyme had been mixed with the substrate and the catalysis began, resulting in a mixture of substrate and product with enzyme. It is unable to confirm that our observation is a substrate-induced or substrate/product-induced dimerization. To solve this, a modified AUC technique, AEC [33], was utilized to detect the quaternary structure change in the absence and presence of substrate (Fig 4). Here the enzyme solution was put into the small well of band-forming centerpiece and then flowed into the substrate-containing channel when the centrifugation began. During the centrifugation, the protein layer gradually sediment and continuously met peptide substrates. Not surprisingly, there was a broad distribution between the monomer and dimer species in the presence of 200 μM peptide substrate, while a major species shifted to the dimer in 400 μM substrate (Fig 4B). It suggests that MERS-CoV Mpro acts as a rapid self-associated and substrate-induced dimerization. Using different strategies, Tomar et al. [14] confirmed that inhibitor binding can also induce and maintain the dimerization of Mpro. On the other hand, we measured the velocity of the product formation during the centrifugation; although the rate (0.017 μM/s) is 10-fold lower than that by the spectrometric assay (Fig 4B).
Fig 4. Charactering the substrate-induced dimerization of MERS-CoV Mpro by AEC [33].

(A) A trace of absorbance at 250 nm for the enzyme in the presence of 200 μM of TQ6-pNA during the experiment. For clarity, only every twice scan and every fifth measuring points per scan are shown. The protein of 15 μg in 10 mM phosphate (pH7.6) was used. The substrate was dissolved in D2O to give a higher density, which can maintain the protein band when the centrifugation begins. The symbols are experimental data and the lines are the results fitted to the Lamn equation using SEDFIT program [31]. The continuous size distribution of the best-fit result was shown in panel B colored by green. (B) Continuous c(s) distributions of MERS-CoV Mpro at the concentration of TQ6-pNA of 0, 200 and 400 μM. The labels M and D showed the position of the monomer and dimer species. (C) Monitoring the enzyme activity of MERS-CoV Mpro during the centrifugation. The same cell in panel A was followed and the absorbance at 405 nm trace for the released product (pNA) was shown. We used the spectra of the first 30 min to calculate the product concentration at different time. The inset plot showed the product versus time and the slope of the line represented the initial velocity.
To quantitatively characterize the monomer-dimer equilibrium of Mpro in the absence and presence of substrate, the AUC results at protein concentrations of 1.5, 6 and 15 μM were globally fitted to the monomer-dimer self-association model (Table 2). The analysis indicated that the Kd value of MERS-CoV Mpro in the absence of substrate was 7.7 μM and this decreased 11-fold in the presence of substrate, which brings it close to the value for SARS-CoV Mpro (1.7 μM; Table 2). Thus it can be concluded that, like the SARS-CoV Mpro R298A mutant [25], the presence of substrate is able to induce the dimerization of MERS-CoV Mpro. In addition, although the Kd values of wild-type SARS-CoV Mpro without or with substrates show no significant difference (Table 2), it was possible to detect substrate-induced dimerization at a protein concentration of 1 μM by AEC [33].
Previous studies have demonstrated that a conserved residue, Glu166, plays a pivotal role in connecting the substrate binding site with the dimerization interface of SARS-CoV Mpro [28]. Here the equivalent residue, Glu169, was mutated and its influence on MERS-CoV Mpro evaluated (Fig 1B). Not surprisingly, compared to the wild-type enzyme, the activity (kcat) of E169A showed a 6-fold decrease (Fig 2B, open diamonds and Table 2). Furthermore, AUC analysis suggested that this enzyme consisted of a single species close to monomeric form even in the presence of substrate (Fig 3E). The Kd values of the E169A mutant without substrate was 14 μM, which is 2-fold higher than that of the wild-type and this did not decrease in the presence of substrate (Table 2). The results suggest that mutation of Glu169 is able to block substrate-induced dimerization and that this results in a decrease in enzyme activity. Unexpectedly, as well as the monomer, some octamer (6.3%) with a sedimentation coefficient of 4.9 S was observed in the presence of substrate (Fig 3E). Previous studies have suggested that a super-active octamer of SARS-CoV Mpro can be locked by 3D domain swapping [35]. In this study, the presence of the octamer form of the MERS-CoV Mpro E169A mutant in the presence of substrate may explain why this mutant is not totally inactive. Taking the above as a whole, the dual role of the conserved Glu residue in catalysis and dimerization is consistent for both Mpro.
Our results confirmed that there are fewer intermolecular polar interactions at the dimerization interface of MERS-CoV Mpro and this results in the enzyme being in the monomer form in aqueous buffer; this contrasts with SARS-CoV Mpro, which is mostly in the dimer form in similar circumstances due to the greater number of intermolecular interactions (Figs 1B and 3B). Based on the sequence alignment, three residues in the dimerization interface vary in coronaviral Mpro sequences (S1 Fig). In SARS-CoV Mpro, residues Arg4, Ser123 and Arg298 are different to the equivalent ones in MERS-CoV Mpro, which are Val4, Thr126 and Met298, respectively. Based on the above, three single mutants, V4R, T126S and M298R, were generated and their proteolytic activity and dimerization were assessed. Unexpectedly, both the V4R and the T126S mutant showed a higher Kd for the dimer to monomer and a lower level of activity than wild-type Mpro (Fig 2B and Table 2). Kd values were decreased by 1.5-fold to 2.4-fold for the two mutants in the presence of substrate, which suggests reduced substrate-induced dimerization (Fig 3C and 3D). Based on the current structure, residue Val4 showed a hydrophobic contact with another protomer’s residue Gly141 (Fig 1B). Mutation of valine to arginine may lose the contact. Furthermore, the side chain of residue Val131 of MERS-CoV Mpro is hydrophobic while that of the equivalent residue, Cys128 of SARS-CoV Mpro, is hydrophilic (Fig 1B). Val131 is close to the Arg4 and will disfavor the electrostatic interaction of Arg4…Glu290 in MERS-CoV Mpro. These variance may result in V4R failed to form a stable dimer. For the T126S mutant, after compared with the other two MERS-CoV Mpro structures (PDB entry 4YLU and 4WME), we found that the side chain of Thr126 is free of rotation. It is able to make a hydrophobic interaction with the residue Tyr121, resulting in a 180°-rotation of the phenol ring, and lead to a hydrogen-bond with the backbone amide of Leu144 (Fig 1B). It further results in the side chain of Leu144 toward another protomer’s Ile300 and make a hydrophobic contact for the two protomers. Mutation of Thr126 to serine will lose this contact and may disfavor the dimerization.
By way of contrast, the M298R mutant resulted in a stable dimer form in phosphate buffer (Fig 3F). The Kd value for the mutant in the absence and presence of substrate were 1.1 and 0.7 μM, respectively, which are very close to those for SARS-CoV Mpro (Table 2). Furthermore, the stable dimer form showed higher proteolytic activity and the mutation transformed the enzymes rate constant pattern at various substrate concentrations into a classical saturation curve (Fig 2C, open triangles). In addition, the Km of the M298R mutant is 5.8-fold lower than that of SARS-CoV Mpro, which suggests a higher substrate binding affinity (Table 2). It can be concluded that mutation of residue Met298 to arginine within the MERS-CoV Mpro results in the stabilization of the dimer formation, which in turn gives rise to more efficient catalysis. Based on our structure, Arg298 is able to make a hydrogen bonding interaction with Thr126. Residue Thr126 can be replaced by serine because the T126S/M298R double mutant also shows similar dimerization characteristics (Fig 3G) and saturated catalytic pattern to that of the M298R mutant (Fig 2C, open hexagons and Table 2). However, it can only be achieved in the presence of Arg298, not Met298.
Substrate binding analysis of MERS-CoV Mpro by ITC
The sigmoid nature of the curve describing the rate constant pattern at various substrate concentrations (Fig 2B) means that it is not possible to obtain a Km value for this enzyme, which would allow us to evaluate the substrate binding affinity of the wild-type MERS-CoV Mpro and its mutants; the exceptions being the M298R single mutant and the T126S/M298R double mutant (Fig 2). To further delineate the binding of substrate to the enzyme, ITC was used to measure the Kd for the substrate (or substrate/product)-enzyme complex and the binding stoichiometry (N) (Fig 5). During the titration, the enzymatic hydrolysis might produce additional heat, resulting in higher ΔH. So we can only compared the N and Kd of the wild-type Mpro with those of E169A and M298R mutants. The three enzymes exhibited similar N (0.89 to 1.06) and Kd (14.2 to 20.3 μM). This suggested that the monomeric and dimeric Mpro show quite the same substrate binding affinity. Such phenomenon is also found in monomeric and dimeric SARS-CoV Mpro, whose N and Kd for the same substrate were 0.97 to 1.05 and 29.9 to 33.8 μM, respectively [28]. With the Km ((k-1+kcat)/k1), kcat and Kd (k-1/k1), we are able to calculate the k1 and k-1, the rate of the association and dissociation of the enzyme-substrate complex. The k1 and k-1 for the M298R mutant and substrate is 0.049 s-1μM-1 and 0.98 s-1, while those for SARS-CoV Mpro is 0.00246 s-1μM-1 and 0.084 s-1. The rate constants for M298R mutant showed 20- and 12-fold higher than those for SARS-CoV Mpro. The more rapid association and relatively slower dissociation of enzyme-substrate complex may be used to explain why the catalytic efficiency (kcat/Km) of the MERS-CoV Mpro M298R mutant is higher than that of SARS-CoV Mpro (Table 2).
Fig 5. ITC titration of peptide substrate binding to MERS-CoV Mpro wild-type (A), E169A (B) and M298R (C) mutants.

Raw data are shown in the top panel of each panel and represent the power input into the sample ampoule over time. Integrated data are shown in the bottom panels in terms of the total energy required for equilibration as a function of the molar ratio of substrate to Mpro. The solid circles show the observed values and the lines represent the fitted results by ligand binding analysis using the Digitam program (TA instruments). The best-fit parameters for stoichiometry (N) and Kd are shown in the bottom panels.
Conclusion
The crystal structure of authentic N-terminus MERS-CoV Mpro was determined and this was found to involve a dimeric form with less intermolecular polar interactions. Biochemical and AUC studies indicated that MERS-CoV Mpro shows almost the same proteolytic activity as SARS-CoV Mpro; although it is a monomer in aqueous buffer and displays substrate-induced dimerization (Fig 6). A conserved residue, Glu169, plays an essential role in the substrate-induced dimerization of both MERS-CoV Mpro and SARS-CoV Mpro. Moreover, mutation of a residue in the dimerization interface, M298R, was found to result in a more stable dimer form in aqueous buffer that had higher enzyme activity; while other two mutations, V4R and T126S, showed the reverse effect. Critical assessment of the residues important to dimerization of and catalysis by MERS-CoV Mpro provides valuable insights into the mechanism that controls the monomer-dimer switch of important and valuable enzyme.
Fig 6. Schematic model of the catalytic process of MERS-CoV Mpro.

Substrate binding results in an active protomer (square) and then dimer formation. After a catalytic cycle, the dimer will dissociate to form the inactive protomers (circle). The M298R mutant, like SARS-CoV Mpro, is able to maintain a dimer form without dissociation before the next round’s substrate binding and catalysis.
Supporting Information
Modified from an output from ESPript [36]. The green ovals indicate the catalytic dyad, while the blue ovals indicate the residues making intermolecular polar contact in the dimer interface of SARS-CoV Mpro. Magenta oval indicate the residue Glu playing dual role for the dimer interface and substrate binding site. Accession numbers are as follows: MERS-CoV, NC_019843.2; Bat-CoV_HKU4, ABN_010865.1; HCoV_HKU1, NC_006577.2; MHV_A59, NP_068668.2; SARS-CoV, NP_828863.1.
(PDF)
Protein identification by SDS-PAGE. M: molecular marker. Lane 1–5: cytoplasmic fraction, flow-through, elute from the nickel affinity column, flow-through after 4-h’s PLpro treatment and protein fraction from S-300 gel-filtration column.
(PDF)
(A) An overlay of the current structure of MERS-CoV Mpro (cyan and orange) with that of SARS-CoV (grey; PDB entry 1uk3). The red arrows show the orientation change affecting the two domain IIIs. Spheres show the two catalytic dyads. (B) Overlay of the current structure with ligand-bound complex (red; PDB entry 4YLU), dimeric C148A mutant (magenta; PDB entry 4WME) and bat-CoV HKU4 Mpro (yellow; PDB entry 2YNA).
(PDF)
The plot of rate constant (kobs) versus the concentration of TQ6-pNA are indicated. The line represented the best-fit results according to the Michaelis-Menten equation (Eq 1). The protein concentration was 1.1 μM. The assays were performed in 10 mM phosphate (pH7.6) and repeated twice to ensure reproducibility and the error bars were shown. The kinetic parameters are shown in Table 2.
(PDF)
The protein concentration of 1.5 (solid line), 6 (dotted line) and 15 μM (dashed line) of MERS-CoV Mpro were used and monitored the size distribution by AUC. The best-fit results suggest that the major species was a monomer with minor shift of sedimentation coefficient (1.74 to 1.97), while the calculated molar mass was from 35.8 to 36.6 kDa. The residual bitmaps of the raw data and the best-fit results are shown in the insets.
(PDF)
(A) Continuous c(s) distribution of the enzyme at peptidyl substrate (TQ6-pNA) concentrations of 0 μM (solid circles), 80 μM (open circles), 200 μM (solid triangles), 340 μM (open triangles), 400 μM (solid squares) and 600 μM (open squares). The protein concentration was 0.25 mg/ml. (B) Sedimentation coefficient shifts of the major species of MERS-CoV Mpro at different TQ6-pNA concentrations.
(PDF)
Acknowledgments
This research was supported by grants from Ministry of Science and Technology, Taiwan (103-2320-B-010-025 and 104-2320-B-010-034) to CYC. We also thank NYMU for its financial support (Aim for Top University Plan from Ministry of Education). Portions of this research were carried out at the National Synchrotron Radiation Research Center, a national user facility supported by the Ministry of Science and Technology of Taiwan, ROC. The Synchrotron Radiation Protein Crystallography Facility is supported by the National Core Facility Program for Biotechnology.
Abbreviations
- 1AUC
analytical ultracentrifugation
- AEC
active enzyme centrifugation
- CoV
coronavirus
- ITC
isothermal titration calorimetry
- MERS-CoV
Middle East respiratory syndrome coronavirus
- Mpro
main protease
- SARS-CoV
severe acute respiratory syndrome coronavirus
- TQ6-pNA
TSAVLQ-para-nitroanilide
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by grants from Ministry of Science and Technology, Taiwan (103-2320-B-010-025 and 104-2320-B-010-034) to CYC. We also thank National Yang-Ming University for its financial support (Aim for Top University Plan from Ministry of Education). Portions of this research were carried out at the National Synchrotron Radiation Research Center, a national user facility supported by the Ministry of Science and Technology of Taiwan, ROC. The Synchrotron Radiation Protein Crystallography Facility is supported by the National Core Facility Program for Biotechnology.
References
- 1. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–20. Epub 2012/10/19. 10.1056/NEJMoa1211721 . [DOI] [PubMed] [Google Scholar]
- 2. Chan JF, Li KS, To KK, Cheng VC, Chen H, Yuen KY. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J Infect. 2012;65(6):477–89. Epub 2012/10/18. 10.1016/j.jinf.2012.10.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson LJ, Baric RS. Emerging human coronaviruses—disease potential and preparedness. N Engl J Med. 2012;367(19):1850–2. Epub 2012/10/19. 10.1056/NEJMe1212300 . [DOI] [PubMed] [Google Scholar]
- 4. Eckerle I, Muller MA, Kallies S, Gotthardt DN, Drosten C. In-vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East Respiratory Syndrome (MERS) Coronavirus infection. Virol J. 2013;10:359 Epub 2013/12/25. 10.1186/1743-422X-10-359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Assiri A, McGeer A, Perl TM, Price CS, Al Rabeeah AA, Cummings DA, et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med. 2013;369(5):407–16. Epub 2013/06/21. 10.1056/NEJMoa1306742 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reusken CB, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, Meyer B, et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis. 2013;13(10):859–66. Epub 2013/08/13. 10.1016/S1473-3099(13)70164-6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis. 2013;19(10):1697–9. 10.3201/eid1910.130946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014;101:45–56. 10.1016/j.antiviral.2013.10.013 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503(7477):535–8. 10.1038/nature12711 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kilianski A, Mielech AM, Deng X, Baker SC. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J Virol. 2013;87(21):11955–62. Epub 2013/08/30. 10.1128/JVI.02105-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin MH, Chuang SJ, Chen CC, Cheng SC, Cheng KW, Lin CH, et al. Structural and functional characterization of MERS coronavirus papain-like protease. J Biomed Sci. 2014;21(1):54 Epub 2014/06/06. 10.1186/1423-0127-21-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chou CY, Lai HY, Chen HY, Cheng SC, Cheng KW, Chou YW. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr D Biol Crystallogr. 2014;70(Pt 2):572–81. Epub 2014/02/18. 10.1107/S1399004713031040 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng KW, Cheng SC, Chen WY, Lin MH, Chuang SJ, Cheng IH, et al. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antiviral Res. 2015;115:9–16. 10.1016/j.antiviral.2014.12.011 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomar S, Johnston ML, St John SE, Osswald HL, Nyalapatla PR, Paul LN, et al. Ligand-induced dimerization of MERS coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals. J Biol Chem. 2015. 10.1074/jbc.M115.651463 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Needle D, Lountos GT, Waugh DS. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 5):1102–11. 10.1107/S1399004715003521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci U S A. 2003;100(23):13190–5. 10.1073/pnas.1835675100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–7. 10.1126/science.1085658 . [DOI] [PubMed] [Google Scholar]
- 18. Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH, Chang GG. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43(47):14958–70. 10.1021/bi0490237 . [DOI] [PubMed] [Google Scholar]
- 19. Hsu MF, Kuo CJ, Chang KT, Chang HC, Chou CC, Ko TP, et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem. 2005;280(35):31257–66. 10.1074/jbc.M502577200 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515–27. 10.1128/JVI.02114-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fan K, Wei P, Feng Q, Chen S, Huang C, Ma L, et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J Biol Chem. 2004;279(3):1637–42. 10.1074/jbc.M310875200 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu A, Wang Y, Zeng C, Huang X, Xu S, Su C, et al. Prediction and biochemical analysis of putative cleavage sites of the 3C-like protease of Middle East respiratory syndrome coronavirus. Virus Res. 2015;208:56–65. 10.1016/j.virusres.2015.05.018 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. [DOI] [PubMed] [Google Scholar]
- 24. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu CG, Cheng SC, Chen SC, Li JY, Fang YH, Chen YH, et al. Mechanism for controlling the monomer-dimer conversion of SARS coronavirus main protease. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 5):747–55. Epub 2013/05/02. 10.1107/S0907444913001315 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–67. Epub 2011/04/05. 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng SC, Chang GG, Chou CY. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys J. 2010;98(7):1327–36. 10.1016/j.bpj.2009.12.4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsieh YH, Chou CY. Structural and functional characterization of human apolipoprotein E 72–166 peptides in both aqueous and lipid environments. J Biomed Sci. 2011;18:4 10.1186/1423-0127-18-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chou YW, Cheng SC, Lai HY, Chou CY. Differential domain structure stability of the severe acute respiratory syndrome coronavirus papain-like protease. Arch Biochem Biophys. 2012;520(2):74–80. Epub 2012/03/07. 10.1016/j.abb.2012.02.015 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320(1):104–24. . [DOI] [PubMed] [Google Scholar]
- 33. Chou CY, Hsieh YH, Chang GG. Applications of analytical ultracentrifugation to protein size-and-shape distribution and structure-and-function analyses. Methods. 2011;54(1):76–82. 10.1016/j.ymeth.2010.11.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chou CY, Chien CH, Han YS, Prebanda MT, Hsieh HP, Turk B, et al. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem Pharmacol. 2008;75(8):1601–9. Epub 2008/03/04. 10.1016/j.bcp.2008.01.005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang S, Zhong N, Xue F, Kang X, Ren X, Chen J, et al. Three-dimensional domain swapping as a mechanism to lock the active conformation in a super-active octamer of SARS-CoV main protease. Protein Cell. 2010;1(4):371–83. 10.1007/s13238-010-0044-8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15(4):305–8. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Modified from an output from ESPript [36]. The green ovals indicate the catalytic dyad, while the blue ovals indicate the residues making intermolecular polar contact in the dimer interface of SARS-CoV Mpro. Magenta oval indicate the residue Glu playing dual role for the dimer interface and substrate binding site. Accession numbers are as follows: MERS-CoV, NC_019843.2; Bat-CoV_HKU4, ABN_010865.1; HCoV_HKU1, NC_006577.2; MHV_A59, NP_068668.2; SARS-CoV, NP_828863.1.
(PDF)
Protein identification by SDS-PAGE. M: molecular marker. Lane 1–5: cytoplasmic fraction, flow-through, elute from the nickel affinity column, flow-through after 4-h’s PLpro treatment and protein fraction from S-300 gel-filtration column.
(PDF)
(A) An overlay of the current structure of MERS-CoV Mpro (cyan and orange) with that of SARS-CoV (grey; PDB entry 1uk3). The red arrows show the orientation change affecting the two domain IIIs. Spheres show the two catalytic dyads. (B) Overlay of the current structure with ligand-bound complex (red; PDB entry 4YLU), dimeric C148A mutant (magenta; PDB entry 4WME) and bat-CoV HKU4 Mpro (yellow; PDB entry 2YNA).
(PDF)
The plot of rate constant (kobs) versus the concentration of TQ6-pNA are indicated. The line represented the best-fit results according to the Michaelis-Menten equation (Eq 1). The protein concentration was 1.1 μM. The assays were performed in 10 mM phosphate (pH7.6) and repeated twice to ensure reproducibility and the error bars were shown. The kinetic parameters are shown in Table 2.
(PDF)
The protein concentration of 1.5 (solid line), 6 (dotted line) and 15 μM (dashed line) of MERS-CoV Mpro were used and monitored the size distribution by AUC. The best-fit results suggest that the major species was a monomer with minor shift of sedimentation coefficient (1.74 to 1.97), while the calculated molar mass was from 35.8 to 36.6 kDa. The residual bitmaps of the raw data and the best-fit results are shown in the insets.
(PDF)
(A) Continuous c(s) distribution of the enzyme at peptidyl substrate (TQ6-pNA) concentrations of 0 μM (solid circles), 80 μM (open circles), 200 μM (solid triangles), 340 μM (open triangles), 400 μM (solid squares) and 600 μM (open squares). The protein concentration was 0.25 mg/ml. (B) Sedimentation coefficient shifts of the major species of MERS-CoV Mpro at different TQ6-pNA concentrations.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.