

Abstract
Purpose
Cancer treatments using tumor defects in DNA repair pathways have shown promising results but are restricted to small subpopulations of patients. The most advanced drugs in this field are Poly(ADP-Ribose) Polymerase (PARP) inhibitors (PARPi), which trigger synthetic lethality in tumors with Homologous Recombination (HR) deficiency. Using AsiDNA, an inhibitor of HR and Non Homologous End Joining, together with PARPi should allow bypassing the genetic restriction for PARPi efficacy.
Experimental design
We characterized the DNA repair inhibition activity of PARPi (olaparib) and AsiDNA by monitoring repair foci formation and DNA damage. We analyzed the cell survival to standalone and combined treatments of 21 tumor cells, and 3 non-tumor cells. In 12 Breast Cancer (BC) cell lines, correlation with sensitivity to each drug and transcriptome were statistically analyzed to identify resistance pathways.
Results
Molecular analyses demonstrate that olaparib and AsiDNA respectively prevent recruitment of XRCC1 and RAD51/53BP1 repair enzymes to damage sites. Combination of both drugs increases the accumulation of unrepaired damage resulting in an increase of cell death in all tumor cells. In contrast, non-tumor cells do not show an increase of DNA damage nor lethality. Analysis of multi-level omics data from BC cells highlighted different DNA repair and cell cycle molecular profiles associated with resistance to AsiDNA or olaparib, rationalizing combined treatment. Treatment synergy was also confirmed with 6 other PARPi in development.
Conclusion
Our results highlight the therapeutic interest of combining AsiDNA and PARPi to recapitulate synthetic lethality in all tumors independently of their HR status.
Keywords: DNA repair, Synthetic lethality, Drug combination, Dbait, PARP inhibitors
Introduction
DNA double-strand breaks (DSBs) are the most lethal of the DNA insults and, if left unrepaired, result in genomic instability and ultimately cell death (1). Therefore, targeted therapies increasing the frequency or the persistence of spontaneous DSBs or DSBs induced by treatments such as radiotherapy or chemotherapy have been extensively studied during the last two decades. The most advanced drugs in this field are the Poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) inhibitors, with clinical trials showing significant benefits in patients with BRCA mutated ovarian cancer (2). Essentially, cells deficient in BRCA1 or 2 are 100- to 1000-fold more sensitive to PARP inhibitors than BRCA1/2 heterozygote or wild-type cell lines (3)(4). PARP is rapidly recruited at site of damage where it strongly auto-modified. The polymers of poly(adenosine diphosphate [ADP]-ribose) formed by PARP are used as a platform for the recruitment of many enzymes involved in Base Excision Repair (BER) (5) and in Microhomology Mediated End Joining (MMEJ) repair of DSBs (6). PARP inhibition prevents BER repair enzymes from being recruited at damage sites (7) and leads to the accumulation of DNA single strand breaks (SSBs) that result in unrepaired stalled replication forks and consequent DSBs. These DSBs are mainly repaired by the Homologous Recombination (HR) repair pathway. Cells with BRCA1/2 mutations are defective in HR (so-called BRCAness) and die directly or indirectly from unrepaired DSBs (1). Cells with functional HR, accurately and efficiently repair DSBs, and are not sensitive to PARP inhibition. Though PARP inhibitor (PARPi) monotherapy showed promising efficacy and safety profiles in the clinic (8)(9), their major limitations are the necessity of HR deficiency (HRD) and the rapid emergence of resistance. Many tumors that initially responded to PARPi treatments finally relapsed through compensatory mutations restoring the HR activity or stimulating the activity of alternative repair pathways such as the non-homologous end joining (NHEJ) pathway (10)(11).
We have recently developed an original class of DNA repair pathway inhibitor, Dbait (12). AsiDNA, a molecule of Dbait family, consists of a 32 base pair oligonucleotide forming a double helix that mimics a DSB. AsiDNA acts by hijacking and hyper-activating PARP1 (13) and the DNA-dependent protein kinase (DNA-PK) (14) which modify the chromatin and consequently inhibit the recruitment of many proteins involved in the HR and NHEJ pathways at the damage sites (14). This strategy sensitizes tumors to DNA damaging therapies such as radiotherapy and chemotherapy (15)(16)(17)(18). The first-in-human phase I trial, combining AsiDNA to radiotherapy to treat patients with skin metastases from melanoma showed encouraging results, with 30% of complete responses (19). We anticipated that AsiDNA could potentiate PARPi activity in BRCA proficient cells by inhibiting HR and establishing a transient state of BRCAness. However, as both drugs act differently on DNA damage response, the inhibitory activities and the efficacy of the association had to be demonstrated.
To test this combined treatment, we first analyzed the effects in DNA repair of the PARPi olaparib (Ola) and AsiDNA, to check that each drug doesn’t interfere with the DNA repair inhibition activity of the other drug. These analyses were performed in the breast cancer (BC) model. BC is the most common female malignancy, with more than 1.7 million new cases diagnosed each year worldwide (20). Inactivating mutations of BRCA are observed in 8.8 % of all sporadic BC tumors (21) with a prevalence of 30% in the Basal-like/Triple negative subgroup (22). We studied the sensitivities to the two drugs alone or in combination in 21 tumor cell lines including BC cell lines with different BRCA status. We observed a synergistic effect of Ola and AsiDNA in all the tested models regardless of BRCA status. Analysis of multi-level omics data from BC cell lines in the context of comprehensive signaling network maps identified different molecular profiles associated with the sensitivity to AsiDNA or Ola, especially in DNA repair and cell cycle mechanisms, highlighting the rational of combining these two drugs. We also demonstrated that this combination is effective using different PARPi, with no toxicity in non-tumor cells.
Materials and methods
Cell culture, chemicals and AsiDNA molecules
Cell cultures were performed with 4 BRCA deficient BC cells lines (BC227 from Institut Curie, HCC1937, HCC38 and MDAMB436 from ATCC), 8 BRCA proficient BC cell lines (BC173 from Institut Curie, BT20, HCC1143, HCC1187, HCC70, MCF7, MDAMB231 and MDAMB468 from ATCC), 3 non-tumor mammary cell lines (184B5, MCF10A and MCF12A from ATCC), 5 human cervical cancer HeLa cell lines silenced for BRCA1 (HelaBRCA1SX, Tebu-Bio referenced as 00301-00041), for BRCA2 (HelaBRCA2SX, Tebu-Bio referenced as 00301-00028), for PARP1 (HeLaPARP1KD, a kind gift of Vincent Pennanaech, Institut Curie, France) and controls (HeLaCTLSX, Tebu-Bio01-00001, and HeLaCTLKD a kind gift of Vincent Pennanaech, Institut Curie, France), human glioblastoma cell lines MO59K and MO59J (DNA-PKcs deficient), human melanoma cell lines SK28LshCTL and SK28 LshDNA-PKcs, human colorectal cancer cell lines HCT116 WT and HCT116 KU70+/- (heterozygote for KU70 gene), human head and neck cancer cell line Hep2, hematologic cancer cell lines Hut78, IM9 and Jurkat. Cells were grown according to the supplier’s instructions. Cell lines were maintained at 37°C in a humidified atmosphere at 5% CO2.
DT40 Burkitt-lymphoma cells are chicken cells that have been knocked out for different genes as previously described in (23). For this study we used DT40 wild type cells as control (DT40WT), and 4 cells lines respectively knocked out for BRCA1, KU70, TDP1 and PARP1 genes (DT40BRCA1KO, DT40KU70KO, DT40TDP1KO and DT40PARP1KO). The DT40 cells were cultured at 37°C with 5% CO2 in Roswell Park Memorial Institute (RPMI-1640) medium supplemented with 1% chicken serum (Life Technologies, Carlsbad, CA, USA), 10-5 M β-mercaptoethanol, penicillin, streptomycin and 10% fetal bovine serum (FBS).Reagents for cell cultivation were obtained from Gibco Invitrogen.
All PARP inhibitors, AZD-2281 (olaparib), AZD-2461, ABT888 (veliparib), MK-4827 (niraparib), BSI-201 (iniparib), BMN673 (talazoparib) and AG-014699 (rucaparib) were purchased from Medchem express (Princeton, USA) and diluted on DMSO to a stock concentration of 10mM.
AsiDNA is a New Chemical Entity, a 64-nucleotides (nt) oligodeoxyribonucleotide consisting of two 32-nt strands of complementary sequence connected through a 1,19-bis(phospho)-8-hydraza-2-hydroxy-4-oxa-9-oxo-nonadecane linker with a cholesterol at the 5′-end and three phosphorothioate internucleotide linkages at each of the 5′ and the 3′ ends (Agilent, USA). The sequence is: 5’- X GsCsTs GTG CCC ACA ACC CAG CAA ACA AGC CTA GA L - CLTCT AGG CTT GTT TGC TGG GTT GTG GGC AC sAsGsC -3’, where L is an amino linker, X a Cholesteryl tetraethyleneglycol, CL a Carboxylic (Hydroxyundecanoic) Acid Linker and s a Phosphorothioate linkage.
Measurement of cellular sensitivity to drugs
AsiDNA or PARPi cytotoxicity was measured by relative survival and cell death quantification. Adherent cells were seeded in 24-well culture plates at appropriate densities and incubated 24hours at 37°C before AsiDNA and/or PARPi addition. Cells were harvested day 6 after treatment, stained with 0.4% trypan blue (Sigma Aldrich, Saint-Louis, USA) and counted with a Burker chamber. Cell survival was calculated as ratio of living treated cells to living mock-treated cells. Cell death was calculated as the number of dead cells on the total number of counted cells. Additivity of the toxicity was calculated by the product of cell survivals to AsiDNA and cell survivals to PARPi.
To measure cytotoxicity in DT40 chicken lymphoma repair mutants (23), 750 cells were seeded in 96-well white plate (final volume 150 μl/well) from Perkin Elmer Life Sciences (Waltham, MA, USA) in media with or without the indicated concentrations of the drugs (AsiDNA and/or veliparib) at 37°C. After 72h, cells were assayed in triplicates with the ATPlite 1-step kit (PerkinElmer, Waltham, MA, USA). Briefly, ATPlite solution was added to each well (150 μl for DT40 cells). After 5min treatment, luminescence intensity was measured by Envision 2104 Multilabel Reader from Perkin Elmer Life Sciences (Waltham, MA, USA). Signal intensities of untreated cells were set as 100%.
Antibodies and immunological studies
For immunostaining, cells are seeded on cover slips (Menzel, Braunschweig, Germany) at a concentration of 5×105 cells and incubated at 37°C during 1 day. Cells are then treated with 16μM AsiDNA +/- 1μM olaparib. 24h after treatment, cells are fixed for 20min in 4% paraformaldehyde/Phosphate-Buffered Saline (PBS 1x), permeabilized in 0.5% Triton X-100 for 10min, blocked with 2% bovine serum albumin/PBS 1x and incubated with primary antibody for 1h at 4°C. All secondary antibodies were used at a dilution of 1/200 for 45min at Room Temperature (RT), and DNA was stained with 4’, 6-diamidino-2-phenylindole (DAPI). The following antibodies were used: primary monoclonal mouse anti-phospho-H2AX (Millipore, Guyancourt, France), anti-53BP1 rabbit antibody (Cell signaling technology, Danvers, USA), anti-Rad51 rabbit antibody (Merk Millipore, Darmstadt, Allemagne), secondary goat anti-mouse IgG conjugated with Alexa-633 (Molecular Probes, Eugene, OR, USA) and secondary goat anti-rabbit IgG conjugated with Alexa-488 (Molecular Probes, Eugene, OR, USA).
Alkaline Single-cell electrophoresis “COMET Assay”
Cells treated with AsiDNA (16μM), olaparib (1μM) or both were suspended in 0.5% low melting point agarose in culture medium and transferred onto a frosted glass microscope slide precoated with a layer of 0.5% normal melting point agarose. Slides were immersed in lysis solution [2.5 mol/L NaCl, 100 mmol/L EDTA,10 mmol/L Tris, 1% sodium lauryl sarcosinate, 10% DMSO, 1% Triton X-100 (pH 10)] at 4jC for 1h, placed in a electrophoresis tank containing 0.3 mol/L NaOH (pH 13) and 1 mmol/L EDTA for 40 min, electrophoresis for 25 min at 25 V (300 mA), washed with neutral buffer [400 mmol/L Tris-HCl (pH 7.5)], and stained with 20 Ag/mL ethidium bromide. The variables of the “comets” were quantified with the software Comet Assay 2 (Perceptive Instrument). Triplicate slides were processed for each experimental point. The tail moment is defined as the product of the percentage of DNA in the tail and the displacement between the head and the tail of the comet.
Inducing photo-damage
These experiments were performed with a Leica SP5 confocal system, attached to a DMI6000 stand using a 63/1.4 objective, under a controlled environment (37°C, 5% CO2). All records were made using the appropriate sampling frequency (512_512 images, line average of four and zooming set to eight) and an argon laser line (514nm for YFP) adapted to the fluorescent protein of interest XRCC1-eYFP. In the first step, two images were acquired within a period of 2–3 s at a laser energy setting sufficiently low not to induce any photodynamic damage. The 405-nm laser line (diode) was then set to maximum output for 100 ms and focused onto a single spot of constant size (176 nm) within the nucleus to cause a point of photo damage with a reproducible amount of energy. Recruitment of XRCC1-eYFP was then monitored by fluorescence using the same setting as for the pre-damage sequence. Laser damage was induced 24 h after treatment with AsiDNA (16μM), olaparib (1μM) or both. Images were captured at 2s intervals for the following 52s. All images were processed using the freely available software ImageJ complemented with the LOCI bioformat plugin (http://www.loci.wisc.edu/ome/formats.html) to open images generated by the Leica SP5 confocal system. A macro was written to automate data extraction from images. Briefly, it consisted of retrieving two regions of interest (ROI), namely the photodamage spot and the nucleus area excluding the spot, and quantifying the total intensity within these ROIs. The latter was used to correct fluorescence intensity for the observational photobleaching. Intensity within the former ROI was normalized to 1, based on quantifications before photodamage, then plotted against time to get the recruitment kinetics.
High-throughput data sources and analysis
mRNA expression analysis
mRNA expression data for BC cell lines were produced using Human Exon 1.0 ST Affymetrix microarrays. Raw data were RMA normalized and summarized with FAST DB annotation (version 2013_1) (24)(25). Gene expression were log2 transformed and mean centred over all the cell line samples and then grouped into the four groups (AsiDNA sensitive, AsiDNA resistant, olaparib sensitive, olaparib resistant). Each gene was assigned with a score using median expression level across samples of the same group and the data was visualized on ACSN map.
Mutation data analysis
Mutation data sets for BC cell lines were retrieved from COSMIC database v71 (http://cancer.sanger.ac.uk) (26). The frequency of mutations for each gene across cell lines in the same groups (AsiDNA sensitive, AsiDNA resistant, olaparib sensitive, olaparib resistant) was calculated and the data was visualized on ACSN map.
Copy number data analysis
The copy number (CN) values for each gene over the cell lines were assessed by GAP analysis of the data generated on affymetrix Genome Wide SNP Array 6.0 (27) and corrected for ploidy (considering four CB as ‘normal’), considering CN less than 3 as loss and CN more than 5 as gain. Then, each gene was assigned with a score using average copy number across samples of the cell lines in the same group (AsiDNA sensitive, AsiDNA resistant, olaparib sensitive, olaparib resistant) and the copy number variation data was visualized on ACSN map.
Spearman rank correlation study
The correlations were assessed by a leave-one-out (LOO) Spearman rank correlation (26)(28). Multiple correction testing was done with Benjamin-Hochberg method (doi = 10.2307/2346101). All tests were considered as two-sided. For each treatment, a list of correlated genes is ranked by correlation p-value. In order to discover the most unique correlated genes with one treatment, a stepwise p-value selection was used so that no gene whose correlation p-value under the selected p-value were retrieved in both treatment. The unique, non-overlapping set of gene robustly correlated with survival to each one of the drugs is provided. The selected p-value (threshold p-value) determined for the ranked genes included in ACSN (29) is 0.005. The Supplementary figure S3-A illustrates the relationship between the number of overlapping genes and the p-value from LOO Spearman correlation analysis.
Data visualization and analysis in ACSN using web-based NaviCell environment
ACSN (29) uses NaviCell environment to navigate maps (30); to analyze and visualize data in the context of maps (31). The enrichment of the ACSN modules with unique genes robustly correlated with survival to each one of the drugs is calculated in the NaviCell toolbox using the standard hypergeometric test, computing the enrichment p-values (P value <0.02) for ACSN modules.
Statistical analysis
All statistical analysis was performed with a two-tailed Student’s t-test.
Results
Molecular mechanisms underlying the combination of AsiDNA and olaparib
As olaparib (Ola) and AsiDNA are both DNA repair inhibitors acting by inhibiting the recruitment of repair enzymes at damage sites, we first checked that each molecule does not impair the capacity of the other to inhibit recruitment of its targeted repair enzymes. One of the first enzymes to be recruited at damage site after auto-modification of PARP is the X-ray repair cross-complementing protein 1 (XRCC1). As expected, Ola significantly delayed the XRCC1 foci recruitment while AsiDNA did not (Figure 1A, B). The recruitment of XRCC1 was similarly delayed in cells treated with Ola in the presence as in the absence of AsiDNA (Figure 1A, B). AsiDNA binds and activates both PARP and DNA-PK in cells. Activation of PARP revealed by the accumulation of Poly-ADP-Ribose polymers was observed in AsiDNA treated cells but not in Ola treated cells or Ola+AsiDNA treated cells indicating that Ola prevents PARP activation by AsiDNA. Activation of DNA-PK kinase activity by AsiDNA can be easily revealed by the pan-nuclear phosphorylation of the histone H2AX (14). This phosphorylation was observed in 80% of treated cells in the presence as in the absence of Ola (Figure 1C). Pan-nuclear phosphorylation of H2AX is thought to be involved in the inhibition of HR and NHEJ repair enzyme recruitment by AsiDNA (14). Ola induces the accumulation of DSBs revealed by the formation of γH2AX foci that co-localize with 53BP1 and Rad51 foci (Figure 1C). The addition of AsiDNA significantly reduced the formation of 53BP1 or Rad51 foci induced by Ola (Figure 1C, D). To demonstrate that the reduction of Rad51 and 53BP1 foci after AsiDNA is induced by the inhibition of their recruitment at damage sites and not through a reduction of the number of DNA damage, we used single cell alkalin comet assays to monitor the damage in MDAMB231 tumor cells after the different treatments. As suggested by γH2AX foci, Ola treatment induced accumulation of damage over 24 hours while AsiDNA did not (Figure 1E). Combining AsiDNA to Ola resulted in a two-fold increase of DNA damage induced by Ola. In MCF10A non-tumor cells, Ola induced formation of few foci of 53BP1 and Rad51 which decreased in cells receiving both Ola and AsiDNA (Figure 1F, G). However in contrast to tumor cells, the non-tumor mammary cells did not show any significant increase of spontaneous damage after treatments with single or combined drugs (Figure 1H).
Figure 1. Effect of the combined treatment AsiDNA and olaparib on DNA repair.
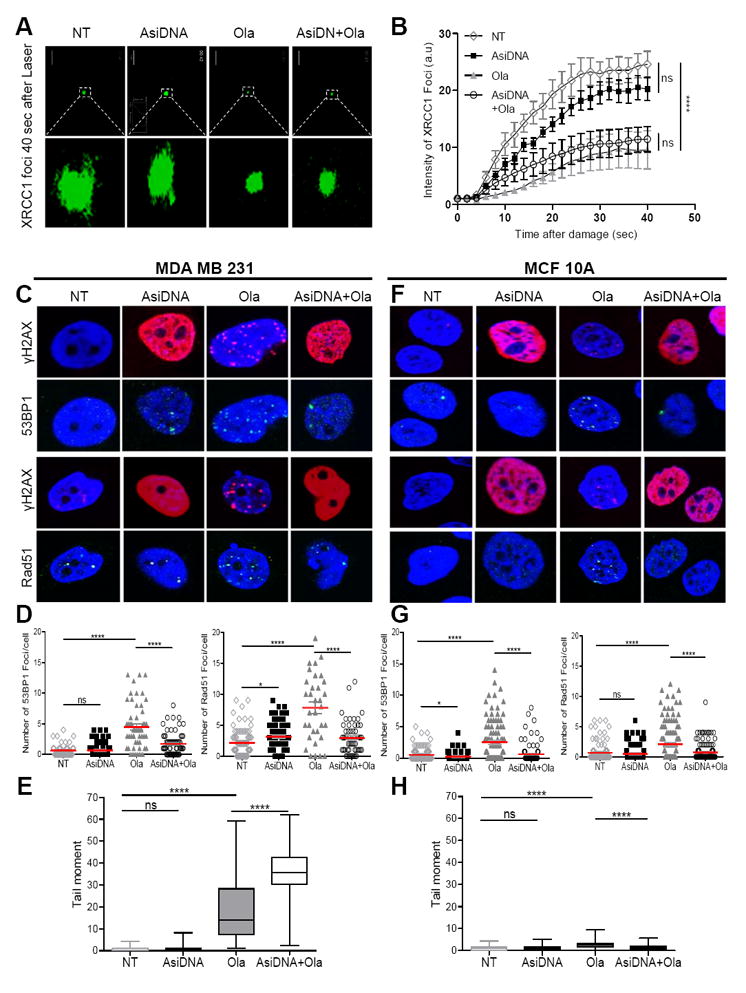
MDAMB231 tumor cells (A, B, C, D, E) and MCF10A non tumor cells (F, G, H) were treated by Ola (1μM) and/or AsiDNA (16μM) for 24 hours. (A, B) cells were damaged by laser irradiation before measuring XRCC1-eYFP repair protein recruitment (A: representative images of recruitment 40 secondes after laser damage; B: kinetics of XRCC1-eYFP recruitment); (C, D, E, F, G, H) Cells were analysed at the end of Ola and AsiDNA treatment for γH2AX (red) and Rad51 or 53BP1 (green) foci formation (C, F : typical nucleus pictures; D, G: quantification in 100 cells for each condition, red bars represent the mean values) and DNA damage using alkaline comet assay (E, H); ns, not significant; *, p<0.05; ****, p<0.0001.
AsiDNA increases olaparib efficacy in cancer cell lines
Efficacy of olaparib (Ola) and AsiDNA was assessed by measuring cell death and proliferation in 21 different cancer cell lines including glioblastoma, cervical cancer, colon cancer, blood cancer, melanoma and breast cancer. The concentration of the drugs (0.1 μM for Ola and 4.8μM for AsiDNA) were chosen based on the 65-75% survival in the BRCA mutant cell lines (Table 1). All tumor models show supra-additive efficacy of the drug combination (Table 1). Moreover, analysis of isogenic pairs with DNA repair mutants to single and combined treatments indicates that AsiDNA is highly cytotoxic to all mutants with one repair defect (PARP1, BRCA1, BRCA2, Ku70, DNA-PKcs) whereas Ola sensitivity is essentially restricted to the BRCA mutants (Table 1). The sensitivity of PARP1, BRCA and Ku70 mutants to AsiDNA was confirmed in an isogenic set of DT40 chicken lymphoma repair mutants (Supplementary figure S1-A), where the highest sensitivity was observed in PARP1 knock-out DT40 cells. As expected, in these cells as in Hela-PARP1 silenced human cells, addition of PARPi did not increased sensitivity to AsiDNA (Table 1 and supplementary figure S1-B). All the other 17 tested solid tumor derived cell lines show a supra-additive response to the combined treatment indicating a suppra-additivity between both inhibitors. In contrast, two out of three of the blood cancer cell lines, Hut78 and Jurkat, had a survival to combined treatment close to the calculated additive effect of both single treatments (Table 1). Taken together, these results indicate that AsiDNA sensitize most tumor cell lines to Ola independently of their BRCA status or other genetic defects.
Table 1. Efficacy of the single and combined treatments in various cancer types.
| Cell line | Tissue | DNA Repair defects | Survival (%/NT)
|
|||
|---|---|---|---|---|---|---|
| Ola | AsiDNA | AsiDNA + Ola | Calculated additivity | |||
| HeLa CTL KD | Cervix | - | 97.2 | 69.0 | 35.4 | 67.1 |
| HeLa PARP1 KD | PARP1 | 100.5 | 49.3 | 47.6 | 49.5 | |
| HeLa CTL SX | - | 105.1 | 96.4 | 79.5 | 101.3 | |
| HeLa BRCA1 SX | BRCA1 | 71.4 | 64.6 | 27.9 | 46.2 | |
| HeLa BRCA2 SX | BRCA2 | 65.0 | 69.3 | 15.1 | 45.1 | |
|
| ||||||
| Hep2 | Head and neck | - | 101 | 69.4 | 42.5 | 70 |
|
| ||||||
| MO59K | Brain | - | 117.3 | 75.4 | 28.7 | 88.5 |
| MO59J | DNA-PKcs | 87.5 | 51.2 | 21.3 | 44.9 | |
|
| ||||||
| SK28 Lsh CTL | Skin | - | 80.6 | 70.3 | 34.8 | 56.7 |
| SK28 Lsh DNA-PKcs | DNA-PKcs | 80.9 | 50.8 | 33.6 | 41.1 | |
|
| ||||||
| HCT116 | Colon | - | 82.9 | 80.0 | 36.6 | 66.3 |
| HCT116 KU70+/- | KU70 | 88.6 | 77.3 | 42.3 | 68.5 | |
|
| ||||||
| Hut78 | Blood | - | 85.8 | 51.1 | 39.1 | 43.8 |
| IM9 | - | 84.6 | 20.3 | 4.6 | 17.1 | |
| Jurkat | - | 68.4 | 45.2 | 28.5 | 31.0 | |
|
| ||||||
| MDAMB231 | Breast | - | 90.9 | 87.2 | 33.5 | 79.2 |
| BC173 | - | 94.4 | 54.5 | 39.3 | 51.4 | |
| BC227 | BRCA2 | 75.3 | 69.8 | 42.6 | 52.6 | |
| HCC38 | BRCA1 | 66.7 | 69.5 | 25.7 | 46.3 | |
| HCC1187 | - | 61.5 | 103.8 | 41.5 | 63.9 | |
| MDAMB468 | - | 68.3 | 53.7 | 28.0 | 36.6 | |
|
| ||||||
| MCF10A | Breast - non tumor | - | 86.2 | 91.7 | 87.5 | 79.0 |
| MCF12A | - | 88.5 | 97.2 | 90.1 | 86.1 | |
Concentrations used were 4.8μM of AsiDNA and 0.1μM of Ola. Calculated additivity = survival to AsiDNA x survival to Ola.
Non-tumor cell lines are not sensitive to the combined treatment AsiDNA and olaparib
Two immortalized mammary cell lines (MCF10A, MCF12A) were analyzed for their sensitivity to the Ola and AsiDNA drug combination. Interestingly, the survival to combined treatment was not decreased in the non-tumor cells (Figure 2). Increasing the dose of Ola to 1 μM had no significant effect on the normal cells but increased the combined treatment efficacy in the breast cancer tumor cells (Figure 2). The synergistic effect was high (three-time higher than expected additivity) in the MDAMB231 cell line which is insensitive to standalone treatment by AsiDNA (Figure 2). In contrast, the non-tumor cells were resistant to Ola and did not show increased sensitivity after addition of AsiDNA (Figure 2; Table 1). In all cell lines, the decrease in the relative number of cells correlated with an increase in cell death (Figure 2B), indicating that the number of living cells reflects a cytotoxic and not a cytostatic effect. Thus, the combined treatment AsiDNA+Ola is specific to tumor cells with no toxicity in normal cells.
Figure 2. The combined treatment AsiDNA and olaparib displays a supra-additive efficacy.
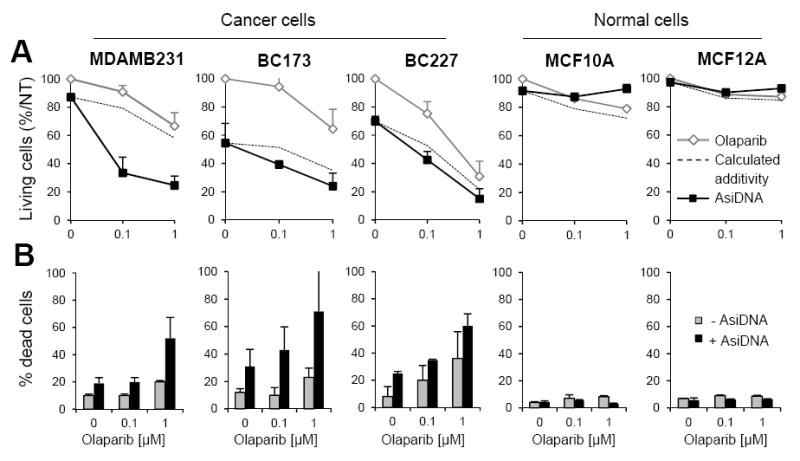
Efficacy of AsiDNA (4. 8μM), olaparib (0, 0.1 and 1μM) or both was monitored 6 days after treatment by cell counting after trypan blue labeling. (A) Percentage of living cells relative to non-treated condition (NT). (B) Percentage of dead cells. Data are expressed as mean + S.D. of at least 6 independent cultures. Dotted lines indicate the calculated cell survivals if additivity between AsiDNA and olaparib.
Analysis of multi-level omics data reveals different profiles of sensitivity to AsiDNA or olaparib in BC cell lines
All the tested cell lines were sensitive to the combined treatment with Ola and AsiDNA suggesting that resistance to both drugs is a very rare event. To better understand this observation, we analyzed the genetic markers associated to resistance to Ola or AsiDNA in a set of 12 BC cell lines (including the 6 BC cell lines tested in combination). No significant correlation between response to AsiDNA and response to Ola was observed in BRCA proficient tumor cell lines (Spearman coefficient r: 0.27 and P value: 0.14; Supplementary figure S2). Only the BRCA-/- cell lines were sensitive to both Ola and AsiDNA single treatments. In order to determine how gene expression profiles could explain the differences in sensitivities to Ola or AsiDNA, we retrieved the “sensitivity” lists of 74 and 71 genes that significantly correlated with sensitivity to respectively AsiDNA or Ola (Supplementary figure S3-A; Supplementary table 1). Interestingly these lists did not display any common gene. Among the genes correlated with sensitivities to AsiDNA or Ola, respectively 9 and 14 genes were directly involved in DNA repair and cell cycle pathways (Table 2).
Table 2. DNA repair and cell cycle genes robustly correlated with survival of BC cell lines to DT01 and olaparib.
| Gene name | Correlation with survival to
|
|
|---|---|---|
| AsiDNA | Olaparib | |
| PPP2R5C | 0.88 | 0.35 |
| CCNA1 | 0.82 | 0.13 |
| FANCE | 0.78 | 0.43 |
| CUL1 | 0.72 | 0.10 |
| MRE11A | 0.69 | 0.25 |
| MAX | 0.67 | 0.31 |
| XRCC1 | -0.63 | 0.19 |
| PPP2R5D | -0.82 | -0.4 |
| AKT3 | -0.83 | -0.4 |
|
| ||
| ATR | 0.41 | 0.82 |
| MBD4 | 0.35 | 0.79 |
| TP53 | -0.32 | 0.78 |
| NEDD4 | 0.15 | 0.77 |
| PRKCH | 0.28 | 0.75 |
| PPP2R5E | 0.42 | 0.75 |
| STAG2 | 0.39 | 0.75 |
| RAD51C | 0.09 | 0.66 |
| ABL1 | 0.32 | -0.51 |
| SMC5 | 0.24 | -0.55 |
| POLB | -0.15 | -0.67 |
| MCPH1 | 0.35 | -0.74 |
| NEIL2 | -0.43 | -0.81 |
| PRKCB | 0.011 | -0.83 |
Numbers indicate correlation coefficients (Spearman r; P value <0.005) between expression of DNA repair and cell cycle-involved genes and survival to AsiDNA or olaparib. (Pale grey: non-significant correlation).
As only the transcriptome was taken into account, the well-known BRCA gene mutations associated to Ola sensitivity were not shortlisted in this analysis. Therefore, we completed the analysis by a multi-level omics data assessment using an Atlas of Cancer Signaling Network resource (ACSN) (29) to integrate mRNA expression, copy number variations and mutational profiles from the BC cell lines. Molecular profiles associated with resistance to AsiDNA or Ola were both quantitatively and qualitatively different and demonstrated that a number of non-overlapping molecular mechanisms are associated with resistance to Ola and AsiDNA (Figure 3; Supplementary figure S4). Interestingly, several molecular mechanisms such as MOMP regulation, Cytoskeleton and Polarity, WNT non-canonical pathway etc. were implicated in response to both drugs, but regulated in an opposite manner (Supplementary Figure S3-B, C, D and Supplementary tables 2 and 3). Cell lines resistant to AsiDNA are characterized by multiple perturbations as expression elevation, copy number gains and mutations in processes involved in cell proliferation, cell survival, EMT and cell motility functional modules (Supplementary Figure S4-A). These data suggest that cells resistant to AsiDNA most likely have a higher proliferation status corresponding to an increase in DNA repair, especially through HR and Fanconi repair pathways (Figure 3A, B). However, cells resistant to Ola show mostly copy number losses in a number of DNA repair pathways, suggesting an opposing function with respect to the Ola response (Figure 3-C, D; Supplementary Figure S4-B). As expected, Ola-sensitive cell lines have an active BER pathway and a defect in HR. In contrast, cells resistant to Ola show multiple losses of copy number in genes involved in the BER pathway, suggesting an inactivation of this pathway. Taken together, omics analyses highlight different molecular mechanisms underlying the response to AsiDNA or Ola, and suggest that repair defects associated to resistance to one drug will increase sensitivity to the other drug making a double resistance very unlikely.
Figure 3. Molecular portraits of sensitivity to AsiDNA or olaparib in BC cell lines on DNA repair and cell cycle map.
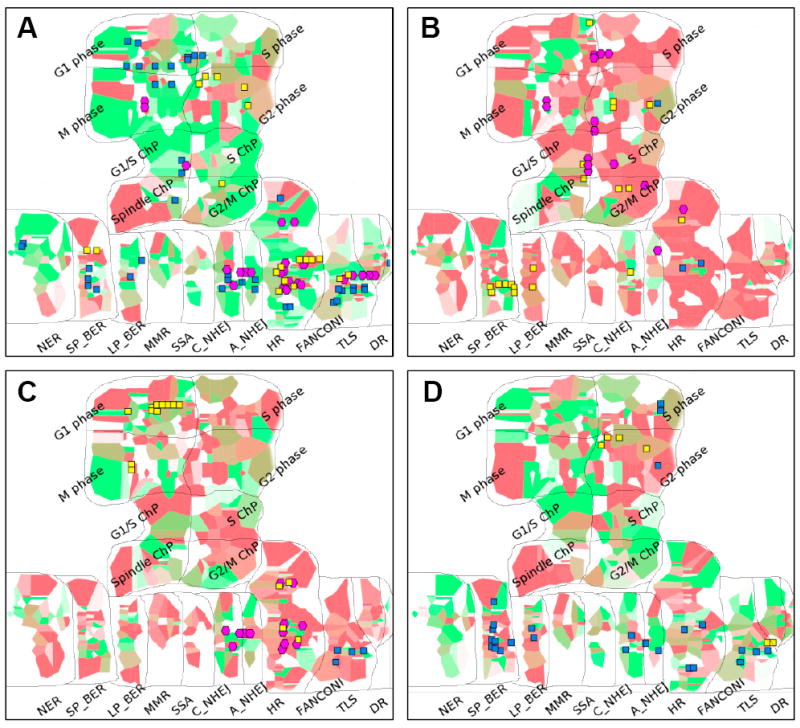
Molecular portraits of BC cell lines according to their sensitivity to AsiDNA (A: sensitive cells; B: Resistant cells) and Ola (C: sensitive cells; D: Resistant cells) using gene expression, copy number variations and mutation frequency, projected on DNA repair and cell cycle map from ACSN. Red and green background colors respectively represent high and low mRNA expression levels across genes of a same pathway; intensity of color shows the level of the change to the mean value of the BC cell lines group. Copy number variations are represented as glyphs where yellow squares indicated copy number gains and blue indicate copy number losses. Mutations are represented using cyan diamonds. Abbreviations: G1/S ChP (G1/S checkpoint), S ChP (S checkpoint), G2/M ChP (G2/M checkpoint), Spindle ChP (Spindle checkpoint); SP_BER (Short-patch BER), LP_BER (Long-patch BER), C_NHEJ (Classical NHEJ), A_NHEJ (Alternative NHEJ).
AsiDNA stimulates efficacy of all PARP inhibitors
PARP inhibitors belong to at least two classes: the catalytic inhibitors that inhibit PARP enzyme activity, and the dual inhibitors that block both PARP enzyme activity and trap PARP proteins on DNA damage sites (23). Ola belongs to the second group whereas veliparib (Veli) is essentially a catalytic inhibitor, as it shows a PARP-trapping activity only at very high doses (23). We repeated the analysis of combination efficacy using Veli instead of Ola (Figure 4A, B). As observed with Ola, the AsiDNA showed a synergistic effect with Veli in the three BC cell lines and not in non-tumor cells. This indicates that trapping PARP on DNA is not essential for an efficient combination.
Figure 4. AsiDNA displays a supra-additive efficacy with different PARP inhibitors.
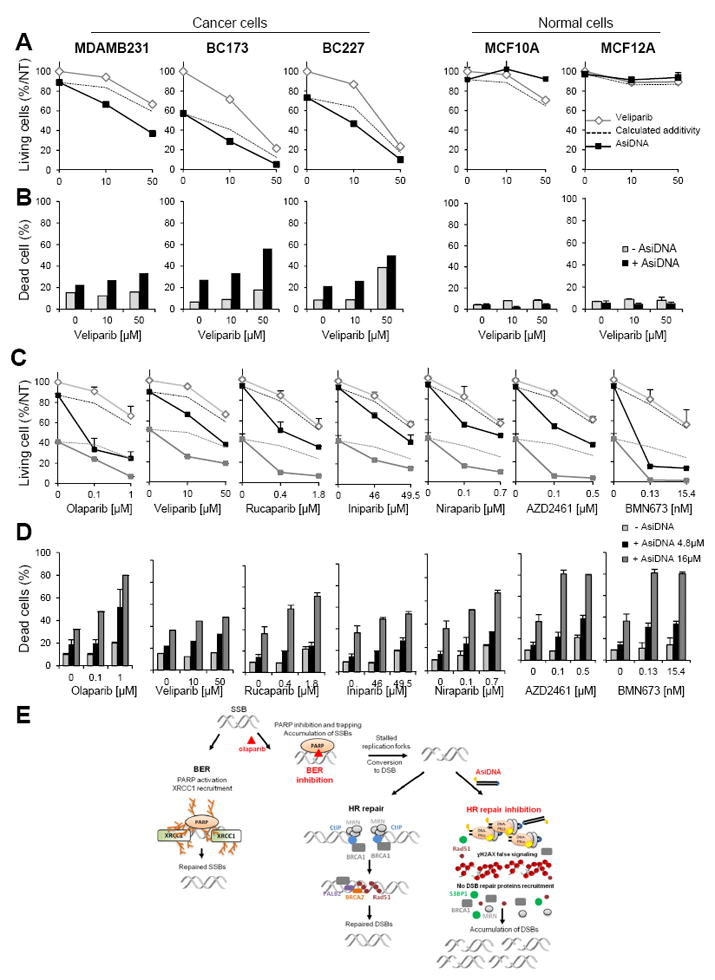
(A, B) Efficacy of AsiDNA (4.8μM), veliparib (0, 10 and 50μM) or both was monitored 6 days after treatment by trypan blue staining. (A) Percentage of living cells relative to non-treated condition (NT). (B) Percentage of dead cells. Dotted lines indicate the calculated cell survivals if additivity between AsiDNA and veliparib. (C, D) Analysis of cell survival (C) and cell death (D) in MDAMB231 cell line in cultures treated with 4.8μM AsiDNA (black), 16μM AsiDNA (dark grey) or not (pale grey). Discontinuous lines indicate calculated cell survivals if additivity between AsiDNA and PARPi (survival to AsiDNA x survival to PARPi). Survivals and cell death were monitored 6 days after treatment. Survivals are expressed as % of living non-treated cells and cell death as frequencies of dead cells. PARPi doses were chosen to give 80% and 50 % survival (Supplementary table 4). (E) Shema of olaparib and AsiDNA repair inhibition mechanisms: 1) BER inhibition by olaparib: inhibition of PARP activity prevents XRCC1 recruitment; 2) HR and NHEJ inhibition by AsiDNA: chromatin modification via pan-nuclear H2AX phosphorylation by activated DNA-PK prevents recruitment of 53BP1, MRN, BRCA1 and RAD51 proteins. Concomitant inhibition of BER, HR and NHEJ is synthetic lethal for cancer cells. SSB, Single-Strand Break; DSB, Double-Strand Break; BER: Base Exision Repair; HR, Homologous Recombination.
We also monitored the efficacy of the combined treatment in MDAMB231 cells with 5 other PARPi (rucaparib, iniparib, niraparib, AZD2461 and BMN673) developed for clinical applications (Figure 4C, D). The applied doses of PARPi were chosen to give a sub-lethal effect and 50% survival (Supplementary table 4). At both doses, the supra-additive efficacy of the combination of PARPi with AsiDNA was confirmed with all the inhibitors (Figure 4C, D) independently of their mechanism of action. These results demonstrate that the observed synergy between AsiDNA and PARPi is a general mechanism and not only restricted to olaparib.
Discussion
Many cancer treatments exploit tumor cell weaknesses or dependencies that are absent or less pronounced, in normal cells. Genetic instability and associated DNA repair defects is one such factor. As some functions become essential during tumor development, molecular networks facilitate compensatory alterations to allow cell survival, a form of “pathway buffering” (32). The idea of targeting tumors with identified genes and proteins that are synthetically lethal with specific tumor suppressor genes (33) has been successfully illustrated by the development of the PARP inhibitors in the treatment of BRCA mutated tumors. However, despite several attempts, only trials which mandate a pathogenic loss-of-function BRCA mutation as an inclusion criterion seemed to give sustained responses (34).
Double-strand DNA breaks are the most lethal DNA lesions and their repair is guaranteed by at least three independent repair pathways that render unlikely the loss of all DSB repair: HR, NHEJ and an alternative Non Homologous End Joining pathway (alt-NHEJ) requiring PARP activity, which takes place when conventional NHEJ and HR fail (35). AsiDNA inhibition of repair enzyme recruitment weakens the ability of the cells to eliminate DSBs. However, as it does not increase the damage occurrence in the cells, AsiDNA toxicity is dependent on spontaneous DNA damage frequency or their induction provoked by associated treatments (14). Interestingly, we observed that Ola significantly increases spontaneous damage frequency. AsiDNA does not induce any damage by itself and therefore do not show any toxicity in cells that do not encounter frequent spontaneous accident or damage as it is the case for non-tumor cells. In these cells, which do not have deregulated repair or cell cycle functions, the AsiDNA addition does not increase the toxicity of PARPi.
Proteins involved in DNA repair and response to damage, such as DSS1, RAD51, NBS1, ATM, ATR, CHEK1, CHEK2 (36), the Fanconi anemia pathway (37), or more recently PTEN (38) have been implicated as possible predictive markers for tumor cell response to PARP inhibitors. Data analysis from BC cell lines in the context of ACSN maps and especially in the context of the DNA repair map, confirms the role of these proteins and their related pathways in Ola sensitivity. Similar analysis on AsiDNA data did not highlight the same molecular mechanisms.
As tumors with “BRCAness”, a profile of tumor associated with deficiency in HR and sensitivity to platinum (39) are relatively rare (21), there is a need to develop new drugs that could recapitulate such features and allow a wider population of patients to benefit from PARPi treatment. The differences in the profile requirement and the lack of toxicity in non-tumor cells make AsiDNA a good candidate for such an association. Essentially, AsiDNA inhibits HR and NHEJ via blinding DNA damage site recognition (12)(14). The general supra-additive effect of the combination of the two drugs indicates independent mechanisms of action. The persistence of the Ola-dependent inhibition of XRCC1 recruitment in the presence of AsiDNA and of the AsiDNA-dependent inhibition of 53BP1 and RAD51 recruitment in the presence of Ola indicate that both drugs act independently. Therefore, Ola inhibits base excision repair resulting in an accumulation of unrepaired SSBs that are converted to DSBs during replication or transcription and cannot be repaired due to the inhibition of HR, NHEJ and altNHEJ by AsiDNA (Figure 4E). The toxicity of PARP inhibitors appears to depend upon the ability of the drug to block PARP at the damage site. We identified that they range in the order of BMN673> AZD2461> Niraparib> Rucaparib> Ola> Iniparib according to their IC20 and IC50 in MDAMB231 (Supplementary table 4). However, all the PARPi showed an enhanced toxicity when administered with AsiDNA. Iniparib, which many believe to not be a bona fide PARPi, as it has very low PARP inhibition in vitro, was only slightly sensitized by AsiDNA. In contrast, Ola and BMN673 were highly potentiated by the addition of AsiDNA. The lack of effect of the combination in non-tumor cells makes the AsiDNA and PARPi combination a potentially interesting treatment in tumors without BRCAness status.
Supplementary Material
Translational relevance.
PARP inhibitors have shown significant benefits in cancer patients with BRCA mutations. However, they show no efficacy in tumors with active homologous recombination repair. In the current study, we propose a novel therapeutic strategy, based on drug combination to achieve synthetic lethality independently of the tumor genetics. We use AsiDNA, a DNA repair pathways antagonist (Dbait molecules family) to deplete double strand break repair activities (homologous recombination and non-homologous end joining) and promote sensitivity to olaparib. The drug driven synthetic lethality is specific to tumor cells and is not observed in non-tumor cells predicting a good safety of the association. As olaparib has obtained FDA approval, and AsiDNA have already been tested in a first-in-man clinical trial, a potential exists for a rapid clinical translation.
Acknowledgments
We thank Nathalie Berthault and Wendy Philippon for their technical participation in this project. We thank the platform RadExp for the comet assays and the LIP for providing the BC227 and BC173 cell lines. We also acknowledge the local CICS platform facilities.
Funding: This work was supported by the SIRIC-Curie, the program PIC SysBio of the Institut Curie, the Centre National de la Recherche Scientifique, the Institut National de la Santé Et de la Recherche Médicale. WJ was supported by a fellowship CIFFRE-ANRT (2013/0907). ST was supported by the Institut National du Cancer (TRANSLA13-081).
Footnotes
Conflict of interest: JSS and MD are cofounders of DNA Therapeutics, the company which holds the patent for AsiDNA.
References
- 1.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001 May;411(6835):366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Gunderson CC, Moore KN. Olaparib: an oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer. Future Oncol. 2015 Jan;11(5):747–57. doi: 10.2217/fon.14.313. [DOI] [PubMed] [Google Scholar]
- 3.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005 Apr;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 4.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005 Apr;434(7035):917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 5.Kubota Y, Nash RA, Klungland A, Schär P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996 Dec;15(23):6662–70. [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015 Jan;6:e1697. doi: 10.1038/cddis.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godon C, Cordelières FP, Biard D, Giocanti N, Mégnin-Chanet F, Hall J, et al. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008 Aug;36(13):4454–64. doi: 10.1093/nar/gkn403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li S. Inhibition of poly(ADP-ribose) polymerase in BRCA mutation carriers. N Engl J Med. 2009 Oct;361(17):1707. doi: 10.1056/NEJMc091621. [DOI] [PubMed] [Google Scholar]
- 9.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013 Aug;14(9):882–92. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 10.Aly A, Ganesan S. BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J Mol Cell Biol. 2011;3(1):66–74. doi: 10.1093/jmcb/mjq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park Y-Y, Kim SM, et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014 Jan;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, et al. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin Cancer Res. 2009 Feb;15(4):1308–16. doi: 10.1158/1078-0432.CCR-08-2108. [DOI] [PubMed] [Google Scholar]
- 13.Croset A, Cordelières FP, Berthault N, Buhler C, Sun J-S, Quanz M, et al. Inhibition of DNA damage repair by artificial activation of PARP with siDNA. Nucleic Acids Res. 2013 Aug;41(15):7344–55. doi: 10.1093/nar/gkt522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quanz M, Chassoux D, Berthault N, Agrario C, Sun J-S, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One. 2009 Jan;4(7):e6298. doi: 10.1371/journal.pone.0006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devun F, Bousquet G, Biau J, Herbette A, Roulin C, Berger F, et al. Preclinical study of the DNA repair inhibitor Dbait in combination with chemotherapy in colorectal cancer. J Gastroenterol. 2012;47(3):266–75. doi: 10.1007/s00535-011-0483-x. [DOI] [PubMed] [Google Scholar]
- 16.Biau J, Devun F, Jdey W, Kotula E, Quanz M, Chautard E, et al. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia. 2014 Oct;16(10):835–44. doi: 10.1016/j.neo.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devun F, Biau J, Huerre M, Croset A, Sun J-S, Denys A, et al. Colorectal Cancer Metastasis: The DNA Repair Inhibitor Dbait Increases Sensitivity to Hyperthermia and Improves Efficacy of Radiofrequency Ablation. Radiology. 2014 Mar;270(3):736–46. doi: 10.1148/radiol.13130805. [DOI] [PubMed] [Google Scholar]
- 18.Herath NI, Devun F, Lienafa M-C, Herbette A, Denys A, Sun J-S, et al. The DNA Repair Inhibitor DT01 as a Novel Therapeutic Strategy for Chemosensitization of Colorectal Liver Metastasis. Mol Cancer Ther. 2016 Jan;15(1):15–22. doi: 10.1158/1535-7163.MCT-15-0408. [DOI] [PubMed] [Google Scholar]
- 19.Le Tourneau C, Dreno B, Kirova Y, Grob JJ, Jouary T, Dutriaux C, et al. First-in-human phase I study of the DNA-repair inhibitor DT01 in combination with radiotherapy in patients with skin metastases from melanoma. Br J Cancer. 2016 May;114(11):1199–205. doi: 10.1038/bjc.2016.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends-An Update. Cancer Epidemiol Biomarkers Prev. 2016 Jan;25(1):16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 21.De Leeneer K, Coene I, Crombez B, Simkens J, Van den Broecke R, Bols A, et al. Prevalence of BRCA1/2 mutations in sporadic breast/ovarian cancer patients and identification of a novel de novo BRCA1 mutation in a patient diagnosed with late onset breast and ovarian cancer: implications for genetic testing. Breast Cancer Res Treat. 2012 Feb;132(1):87–95. doi: 10.1007/s10549-011-1544-9. [DOI] [PubMed] [Google Scholar]
- 22.Greenup R, Buchanan A, Lorizio W, Rhoads K, Chan S, Leedom T, et al. Prevalence of BRCA mutations among women with triple-negative breast cancer (TNBC) in a genetic counseling cohort. Ann Surg Oncol. 2013 Oct;20(10):3254–8. doi: 10.1245/s10434-013-3205-1. [DOI] [PubMed] [Google Scholar]
- 23.Murai J, Huang SN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012 Nov;72(21):5588–99. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003 Apr;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 25.de la Grange P, Dutertre M, Martin N, Auboeuf D. FAST DB: a website resource for the study of the expression regulation of human gene products. Nucleic Acids Res. 2005 Jan;33(13):4276–84. doi: 10.1093/nar/gki738. [Internet] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015 Jan;43(Database issue):D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popova T, Manié E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009 Jan;10(11):R128. doi: 10.1186/gb-2009-10-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martignetti L, Laud-Duval K, Tirode F, Pierron G, Reynaud S, Barillot E, et al. Antagonism pattern detection between microRNA and target expression in Ewing’s sarcoma. PLoS One. 2012 Jan;7(7):e41770. doi: 10.1371/journal.pone.0041770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuperstein I, Bonnet E, Nguyen H-A, Cohen D, Viara E, Grieco L, et al. Atlas of Cancer Signalling Network: a systems biology resource for integrative analysis of cancer data with Google Maps. Oncogenesis. 2015 Jan;4:e160. doi: 10.1038/oncsis.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuperstein I, Cohen DPA, Pook S, Viara E, Calzone L, Barillot E, et al. NaviCell: a web-based environment for navigation, curation and maintenance of large molecular interaction maps. BMC Syst Biol. 2013 Jan;7:100. doi: 10.1186/1752-0509-7-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnet E, Viara E, Kuperstein I, Calzone L, Cohen DPA, Barillot E, et al. NaviCell Web Service for network-based data visualization. Nucleic Acids Res. 2015 Jul;43(W1):W560–5. doi: 10.1093/nar/gkv450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartman JL, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001 Feb;291(5506):1001–4. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- 33.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997 Nov;278(5340):1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 34.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013 Aug;14(9):882–92. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 35.Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen. 2015 Nov;793:166–75. doi: 10.1016/j.mrgentox.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 36.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006 Aug;66(16):8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 37.Venkitaraman AR. A growing network of cancer-susceptibility genes. N Engl J Med. 2003 May;348(19):1917–9. doi: 10.1056/NEJMcibr023150. [DOI] [PubMed] [Google Scholar]
- 38.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim J-S, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009 Sep;1(6-7):315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer. 2004 Oct;4(10):814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.