

Abstract
DNA methylation at the promoter of a gene is presumed to render it silent, yet a sizable fraction of genes with methylated proximal promoters exhibit elevated expression. Here, we show, through extensive analysis of the methylome and transcriptome in 34 tissues, that in many such cases, transcription is initiated by a distal upstream CpG island (CGI) located several kilobases away that functions as an alternative promoter. Specifically, such genes are expressed precisely when the neighboring CGI is unmethylated but remain silenced otherwise. Based on CAGE and Pol II localization data, we found strong evidence of transcription initiation at the upstream CGI and a lack thereof at the methylated proximal promoter itself. Consistent with their alternative promoter activity, CGI-initiated transcripts are associated with signals of stable elongation and splicing that extend into the gene body, as evidenced by tissue-specific RNA-seq and other DNA-encoded splice signals. Furthermore, based on both inter- and intra-species analyses, such CGIs were found to be under greater purifying selection relative to CGIs upstream of silenced genes. Overall, our study describes a hitherto unreported conserved mechanism of transcription of genes with methylated proximal promoters in a tissue-specific fashion. Importantly, this phenomenon explains the aberrant expression patterns of some cancer driver genes, potentially due to aberrant hypomethylation of distal CGIs, despite methylation at proximal promoters.
In mammalian DNA, cytosines within CpG dinucleotides are heavily methylated throughout the genome, yet there are several discrete “islands” that contain a high frequency of unmethylated CpG sites. These are called CpG islands (CGI), and their identification has long been considered important in the annotation of functional landmarks within the genome. Historically, CGIs served as landing strips to locate annotated genes (Larsen et al. 1992), and it was for good reason as it was later discovered that 55%–60% of all genes contain CGIs at their annotated promoters. While about half of all CGIs in the genome coincide with gene promoters, the remaining half are either intragenic or intergenic and are termed “orphan CGIs” due to their remote location that suggested the uncertainty over their biological significance (Deaton and Bird 2011).
Does there exist evidence to support the idea that orphan CGIs are involved in gene regulation? Indeed, several specific examples, showing promoter activity at orphan CGIs, were uncovered in the context of critical functions like imprinting and development (Deaton et al. 2011). For example, a CGI in intron 10 of the imprinted Kcnq1 gene (Mancini-DiNardo et al. 2003) promotes the initiation of a noncoding transcript (Kcnq1ot1) required for the imprinting of several genes at this locus. Tissue-specific alternative promoter activity was detected at another orphan CGI that promotes a specific isoform of the Rapgef4 gene (Hoivik et al. 2013). Cumulative evidence suggests that most, perhaps all, CGIs have promoter-like characteristics and are sites of transcription initiation (Illingworth et al. 2010). Additionally, most of the conserved methylation differences between tissues occurred at orphan CGIs (Illingworth and Bird 2009), suggesting that they are tightly regulated. A recent study that derived CGI annotations from experimental methylation data (eCGIs) also showed that promoter-distal eCGIs exhibited the most tissue-specific methylation patterns and were linked to the tissue-specific production of alternative transcripts (Mendizabal and Yi 2015). In fact, studies profiling CpG methylation patterns have identified differentially methylated regions (DMRs) even in the shores of CpG islands (Pollard et al. 2009). These regions of lower CpG density in close proximity (up to 2 kb) to CGIs, whose differential methylation patterns are strongly related to gene expression, are highly conserved and have distinct tissue-specific methylation patterns (Irizarry et al. 2009a). Thus, over time, CG-dense genomic loci (i.e., CGIs and their shores) have been realized to be increasingly important in many functional contexts, and their immense regulatory potential outside of annotated promoters is only beginning to be understood.
Typically, methylation at a gene's promoter renders it silent (Han et al. 2011) by modifying DNA accessibility to the transcriptional machinery (Suzuki and Bird 2008) or by recruiting factors that aid in generating a refractory chromatin conformation unsuitable for transcription (Kouzarides 2007). While several prior studies (Suzuki and Bird 2008; Deaton and Bird 2011; Sproul et al. 2011; Smith et al. 2012; van Eijk et al. 2012) have observed strongly negative correlations between promoter methylation and gene expression, others report more nuanced relationships between the two (Shilpa et al. 2014; Wagner et al. 2014; Martino and Saffery 2015; Wan et al. 2015), including a lack thereof. Additionally, there are several instances of genes in cancer cells wherein abnormal expression is persistent despite widespread promoter hypermethylation (Van Vlodrop et al. 2011; Guillaumet-Adkins et al. 2014; Moarii et al. 2015). These collectively indicate that additional factors controlling expression of genes with methylated promoters have not been identified. Furthermore, due to the longstanding interest in CGI-promoter genes, most of this knowledge is based on analysis of CGI-promoters (Jones 2012), and the details of the role of methylation in controlling non-CGI transcription start sites (TSSs) have largely been overlooked.
This lack of consensus prompted us to explore the global transcriptional landscape of methylated-promoter genes. We found that substantial numbers of methylated-promoter genes (∼1500 in each of 34 tissues) are expressed at high levels; such promoters are predominantly non-CGI, which is consistent with prevailing knowledge on the rarity of methylation at CGI promoters (Brandeis et al. 1994; Illingworth et al. 2010; Lienert et al. 2011). While the expression of many such genes can be attributed to the use of alternate gene body promoters, as has been shown in some normal and cancer cells (Maunakea et al. 2010; Nagarajan et al. 2014), we estimate that the high levels of expression realized by almost 50% of all methylated-promoter genes remain completely unexplained.
Here, we assessed across 34 primary human tissues and cell types, the extent to which the genes with methylated and silenced promoters utilize an upstream CpG island as an alternative promoter to express their gene product.
Results
Highly expressed genes with methylated promoters
We obtained RNA-seq expression and whole-genome bisulfite sequencing (WGBS) methylation data for 30 primary tissues and four cell lines from the Roadmap Epigenomics Project (Bernstein et al. 2010) and other sources (Supplemental Table 1; Djebali et al. 2012; Ziller et al. 2013; Menafra et al. 2014; Lay et al. 2015). Henceforth, we will refer to these 34 samples simply as “tissue types.” In a given tissue type, there exists, on average, about 9000 genes whose primary promoters are maintained in a heavily methylated state (see Methods). Although methylation at a gene's promoter is expected to render it silent, we observed that ∼1500 of such genes exhibited high levels of expression. We then excluded genes whose expression could be explained by alternative gene body promoter activity (see Methods), and this resulted in 700 genes in each tissue whose expression remains unexplained. To specifically assess the involvement of the closest upstream CGI in the expression of these genes, we restricted downstream analysis to only those genes that did not have another gene annotated (including noncoding RNAs) in the genomic region between the gene's transcription start site and the CGI. This eliminates potential biases owing to intervening transcriptional activity. We further verified that this subset of genes was not enriched for any specific biological function or expression status compared to the set of all methylated-promoter genes (Supplemental Fig. 1). These filters resulted in a set of ∼3200 methylated-promoter genes (down from ∼9000 overall) out of which ∼440 (down from ∼1500) are highly expressed per tissue. The numbers of genes at various filtering stages across tissues are provided in Supplemental Table 2. Additionally, methylated-promoter gene names and their methylation status across tissues are listed in Supplemental Table 3.
In all 34 tissues, we find that a vast majority (∼90%) of these genes do not contain CpG islands in their promoters, which is significant enrichment relative to a 30% expectation of non-CGI promoter genes genome-wide (Saxonov et al. 2006). This result agrees with prevailing knowledge on the rarity of methylated CGIs at the promoters of annotated genes (which is only ∼3% overall) (Illingworth et al. 2010), as well as the lower propensity of CGI promoters to be de novo methylated compared to non-CGI promoters (Brandeis et al. 1994; Lienert et al. 2011). Further, they are enriched for cell-type–specific functions based on a quantitative index of tissue specificity (TSI) (Yanai et al. 2005; see Methods) as well as Gene Ontology (GO) enrichment. Supplemental Figure 2 shows that the median TSI of expressed methylated-promoter genes is significantly greater compared to that of all genes (10−4 < Wilcoxon signed-rank test P < 0.05 in 33/34 tissues showing a significant trend). We also present overrepresented functional terms based on GO enrichment in each tissue in Supplemental Figure 3. These findings are in line with existing knowledge that a majority of widely expressed genes use CpG island promoters, while most tissue-specific genes have neither CpG islands nor TATA-boxes (Larsen et al. 1992; Zhu et al. 2008) in their promoters.
Association of distal CGI with methylated-promoter gene expression
Across the set of all methylated-promoter genes in a given tissue, we asked if the methylation status of the closest upstream CGI was informative of its expression. Specifically, we categorized these genes into two sets—(1) expressed (MethExp), and (2) not expressed (MethNotExp) genes (see Methods)—and compared the proportion of methylated distal CGIs in each case. As shown in Figure 1A, we observe a strong negative relationship between CGI methylation and the corresponding gene's expression (1.25 < Odds ratio < 1.75, 10−10 < Fisher's exact test P < 0.01 in 26/34 tissue types showing a significant trend). We further found that CGIs associated with MethExp genes tend to have significantly lower methylation than those associated with MethNotExp genes (10−13 < Wilcoxon P < 0.02 in 32/34 tissues showing a significant trend) (Fig. 1B). Therefore, we conclude that expression levels of methylated-promoter genes are strongly associated with the epigenetic status of the distal upstream CGIs.
Figure 1.

Association of distal CGI with the expression of MethExp genes. (A) Odds ratio and 95% confidence interval (CI) (y-axis) of the proportion of unmethylated CGIs upstream of MethExp genes versus MethNotExp genes in 34 tissue types (x-axis). A depletion of methylation at CGIs upstream of MethExp genes corresponds to a higher odds ratio. (B–E) Comparison of various properties for the CGIs upstream of MethExp genes (yellow) versus MethNotExp (red) genes; (B) fractional methylation level, (C) genomic distance to gene, (D) phastCons scores, and (E) derived allele frequencies (DAFs). The median and the 95% CI (x-axes) are shown for 34 tissue types (y-axes). (F) Phylogenetic tree of the eight vertebrate species used in determining the extent of shared synteny with human among methylated-promoter genes. The association between CGI-gene synteny and whether the gene is MethExp or MethNotExp was assessed via regression, while controlling for genomic distance between CGI and the gene. The significance of the association (P-value) is shown on each branch corresponding to the species used to estimate synteny with respect to human. Statistically significant associations (P < 0.05) are annotated with an asterisk in all plots.
On average, CGIs upstream of MethExp genes are located at a distance of 10 kb and, interestingly, are several-fold closer to their associated genes than those upstream of MethNotExp genes (10−13 < Wilcoxon P < 10−4 in 33/34 tissues showing a significant trend) (Fig. 1C). While such proximity might not be a prerequisite for intergenic CpG islands to act as alternative promoters to transcribe genes with silenced primary promoters, it does seem likely that it would be a preferred configuration.
Finally, the CGIs associated with MethExp genes are evolutionarily much more conserved than those associated with MethNotExp genes, both between species (using phastCons scores based on an alignment of 46 vertebrates; 10−4 < Wilcoxon P < 0.05 in 27/34 tissues) (Fig. 1D; Siepel et al. 2005) and within species (using average derived allele frequencies [DAF] across humans; 10−7 < Wilcoxon P < 0.05 in 20/34 tissues) (Fig. 1E; see Methods). Additionally, from annotations of syntenic blocks between human and eight related vertebrate species (see Methods), we assessed the extent to which shared synteny between a methylated-promoter gene and its upstream CGI was informed by the expression status of the gene, using a logistic regression framework that controlled for the genomic distance between them. We found that MethExp genes and their upstream CGIs are more often in the same syntenic block than CGIs upstream of all other genes (10−3 > P-value attached to coefficient of expression status > 10−6 in 6/8 comparisons) (Fig. 1F). Interestingly, this holds true only in the six mammalian species and not in either of the two nonmammalian vertebrates, which suggests that MethExp-associated CGIs were only recently co-opted, close to the base of mammalian divergence, to function as alternative promoters. Thus, higher purifying selection acting specifically on MethExp-CGIs that are also in synteny with their associated genes is indicative of their functional role, in this case, as a regulatory element (promoter) facilitating transcription of the downstream gene.
We further hypothesized that the tissue-specific usage of CGIs as alternative promoters may be regulated by cell-type–specific transcription factors (TFs). To test this, for every CGI showing evidence of alternative promoter activity in some cell type, we identified the high confidence TF binding sites (see Methods) in those CGIs and tested if TFs corresponding to these sites show a preference to be expressed in cell types where the CGI was active versus not. Consistent with expectations, a large fraction of these CGIs (∼40% vs. a 5% random null expectation; Fisher's P < 10−16) do show patterns of cell-type–specific regulation.
Transcription initiation occurs at distal CGI and not the promoter of MethExp genes
Our previous observation of lower methylation and increased conservation at CGIs upstream of MethExp genes is only suggestive of their potential to function as alternative promoters to transcribe them. Here, we explicitly test for transcriptional initiation at these CGIs using two different experimental measures.
First, we used single molecule Cap Analysis of Gene Expression (CAGE) data from the FANTOM Consortium (The FANTOM Consortium and the RIKEN PMI and CLST (DGT) 2014), available for 15 tissue types. The CAGE assay produces a snapshot of the 5′ end of the messenger RNA population in a biological sample, which provides a direct quantitative measure of initiation rate at a given locus. Thus, in a tissue-specific fashion, we quantified the transcription initiation signal (number of CAGE tags) at the promoters as well as the associated CGIs of three groups of genes: (1) MethExp; (2) MethNotExp; and (3) expressed genes with methylation-free promoters (NotMethExp). This third group serves as a baseline for the amount of initiation expected at similarly expressed gene loci. Since expression level is related to the intensity of initiation signal, we ensured by sampling that the expression level distribution of the selected MethExp and NotMethExp genes were comparable. Figure 2, A and B, shows the distribution of CAGE levels at the promoters as well as the associated CGIs of MethExp, MethNotExp, and NotMethExp genes pooled across all tissues, respectively. In the case of promoters, we observe that the number of CAGE tags is quite low at MethExp genes and, importantly, is several-fold less than that at similarly expressed NotMethExp genes (Wilcoxon P = 10−6). Further, the complete lack of CAGE tags at MethNotExp genes is consistent with the fact that these genes are not expressed at all. Next, we contrast the transcription initiation signal at upstream CGIs associated with the three gene groups. It is known that most, perhaps all, CGIs are sites of transcription initiation, and it is owing to this property that ∼50% of them are adapted for promoter function and coincide with the TSS of annotated genes (Deaton and Bird 2011). Consistent with this expectation, CGIs from all gene groups show substantial CAGE tag levels. Considering that CGIs associated with MethNotExp genes do not contribute to the expression of those genes, the CAGE level at these CGIs may serve as a baseline expectation for orphan CGIs. Then, interestingly, we observe that the CAGE at CGIs associated with MethExp genes is significantly greater than this baseline (Wilcoxon P = 10−4). In fact, MethExp-associated CGIs collectively exhibit somewhat greater transcriptional activity (CAGE) than even the NotMethExp-associated CGIs (Wilcoxon P = 0.04). Low coverage of CAGE tags at orphan CGIs limits our ability to statistically substantiate comparisons between groups at the per-tissue resolution, but the promoter and CGI CAGE trends we observe across gene groups in the above analyses are consistent in 15/15 and 12/15 tissues, respectively (see Supplemental Fig. 4), and therefore does not affect our conclusion.
Figure 2.
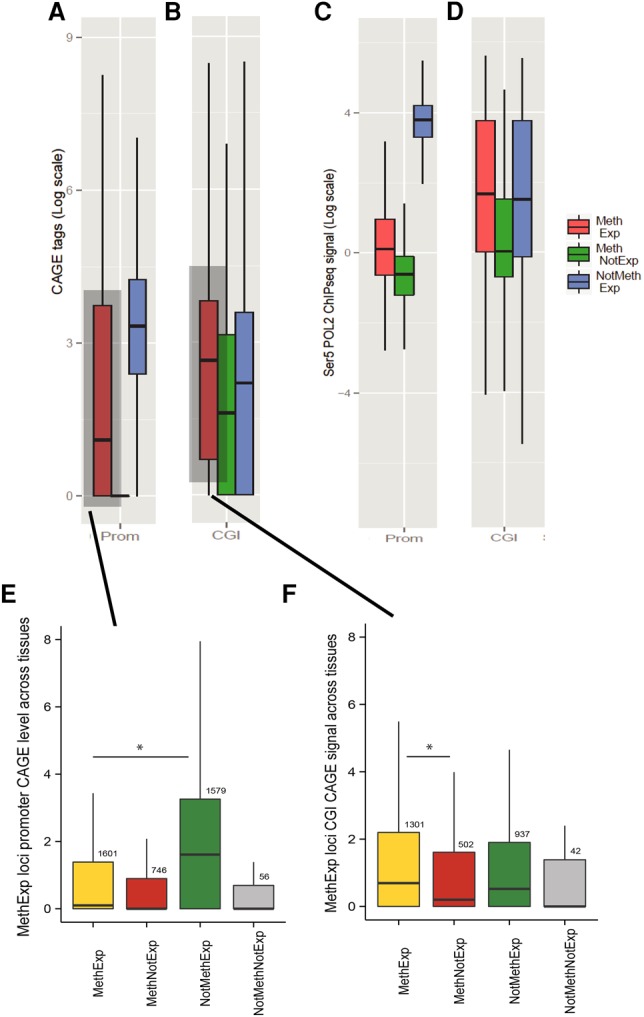
Transcription initiation occurs at an upstream alternative CGI promoter and not at the proximal promoters of MethExp genes. The evidence of transcriptional initiation based on CAGE tag intensity (log-transformed y-axis) is contrasted for three gene groups, MethExp (red), MethNotExp (green) and NotMethExp (blue) at (A) the proximal promoter and (B) the distal CGI (x-axis). C and D are analogous to A and B, respectively, for observed levels of transcription initiation based on Ser5-Pol II ChIP-seq intensity. For the pan-tissue pooled set of MethExp genes, the plot shows the CAGE signal (y-axis) at the (E) promoter and (F) associated distal CGIs of these genes when they are MethExp (yellow), MethNotExp (red), NotMethExp (green), and NotMethNotExp (gray) in other tissues (x-axis).
The second measure we used for the quantification of initiation corresponds to a signal associated with serine-5-phosphorylated RNA Polymerase II (Pol II-Ser5). Specifically, the initiating form of Pol II is phosphorylated at Ser5, and as elongation of the mRNA molecule occurs, the enzyme gradually loses Ser5-P and gains Ser2-P (Phatnani and Greenleaf 2006; Jonkers and Lis 2015). To this end, we used Ser5-P Pol II chromatin immunoprecipitation sequencing (ChIP-seq) data in the MCF-7 cell line (based on data availability) to quantify transcriptional initiation at the promoters (Fig. 2C) and upstream CGIs (Fig. 2D) of the three gene groups. The trends are highly consistent with those obtained from CAGE. Specifically, initiation signal at the promoters of MethExp genes is much lower than NotMethExp genes (Wilcoxon P < 10−5) that are expressed at comparable levels. Also, consistently, CGIs at MethExp have higher Ser5-P signals than MethNotExp genes (Wilcoxon P < 10−5) as well as NotMethExp (albeit not yielding statistical significance; Wilcoxon P = 0.15), suggesting that, specifically for MethExp genes, the upstream CGI may serve as an alternative, hitherto undetected promoter.
In light of the above observations, it is also possible that distal CGIs associated with MethExp genes are in fact their true primary promoters that were misannotated, likely due to the narrow expression breadth (i.e., tissue specificity) of MethExp genes. To distinguish between the alternative scenarios of promoter misannotation and unsuspected context-specific distal promoter usage, we carried out two specific analyses. First, we performed a locus-specific cross-tissue comparison of CAGE tags at the annotated promoters of MethExp genes when they are categorized as MethExp, MethNotExp, and NotMethExp across different tissues (Fig. 2E). If our observations were simply due to misannotation, then specifically for these select group of genes whose promoters become methylated in some tissue, we expect to see a ubiquitous lack of transcription initiation at their annotated promoters across all other tissues, regardless of their methylation status. Instead, we find that the CAGE tags at the annotated promoters of these select genes when they are unmethylated is very high (NotMethExp >> MethExp or MethNotExp; Wilcoxon P < 10−3 in both cases), supporting the idea that MethExp-associated CGIs serve only as alternative promoters, and not the primary ones. Further, a similar locus-specific cross-tissue comparison of CAGE tags at distal CGIs (Fig. 2F) showed that CGIs have higher CAGE tags in tissues where their associated genes are MethExp compared with tissues where they are MethNotExp (Wilcoxon P = 0.008). However, MethExp and NotMethExp groups do not show a significant difference in CAGE levels at the distal CGI, suggesting that transcriptional activity at distal CGIs in these instances is generally unlinked with promoter activity. In addition, we also observe that the promoter methylation levels are starkly different when these loci are active versus silent across tissues (Supplemental Fig. 5).
Next, we assessed the effect of loss of methylation on the relative activity of the annotated promoter of MethExp genes. This analysis is, however, limited by the availability of data in human. We therefore analyzed MethExp genes in mouse embryonic stem cells with (WT; wild-type cells) and without DNA methyl transferase activity (DNMT TKO; DNMT triple knockout cells). Using RNA-seq and WGBS methylation data in WT cells, we identified all high-confidence (about 103) MethExp genes using the same protocol as that for tissue types in human. After verifying that they exhibit the same broad features of CGI alternative promoter use as the MethExp genes in human tissue types (Supplemental Fig. 6), we analyzed their promoter usage patterns in DNMT TKO cells. We hypothesized that, if the distal CGI was the only promoter of these genes, then removing methylation at the annotated promoters should not lead to a change in their activity status. Unlike CAGE, RNA-seq does not allow for direct quantification of transcription initiation at these annotated promoters. Therefore, in DNMT TKO cells, we contrasted the mean read density observed upstream of the annotated TSS (TSS−200 bp) to that observed downstream from it (TSS + 200 bp), relative to the same in WT, for every gene identified as MethExp in WT. We see that 68 out of 103 MethExp genes show an increase in mean read density (normalized by the corresponding densities in WT) downstream from the annotated TSS (Fisher's P = 0.02), hinting at a potential switch from usage of distal CGIs to the annotated promoters in these cases. Note that, in the absence of data in mouse knockouts that directly quantifies initiation rates, we cannot conclusively ascertain that the increased numbers of reads downstream from the TSS in DNMT TKO cells are from transcripts originating at the annotated TSS; this result, therefore, must be considered with caution. However, taken together, we conclude that our overall observations are not simply a reflection of erroneous promoter annotation.
As an additional layer of evidence for transcriptional activity, we quantified the repressive histone modifications (H3K9me3 and H3K27me3) at the promoters of MethExp, MethNotExp, and NotMethExp genes (Supplemental Fig. 7). Consistent with other observed features of active transcription, we find that both of these marks are significantly higher at MethExp than NotMethExp promoters (H3K9me3: 10−69 < Wilcoxon P < 10−5 in 22/32 tissues; H3K27me3: 10−113 < Wilcoxon P < 10−3 in 19/32 tissues). Further, given that distal CGIs can display transcriptional activity similar to promoters, it is likely that they also harbor histone modifications reflective of their activity status. To this end, we contrasted the ChIP-seq signal of two active (H3K4me3, H3K9ac) and two repressive (H3K27me3, H3K9me3) histone modifications at CGIs associated with MethExp, MethNotExp, and NotMethExp genes (Supplemental Fig. 8). In addition, we also compared the DNase hypersensitivity signal to assess the extent of chromatin accessibility (also reflective of transcriptional activity) at these CGIs. The tests for histone marks were performed for different numbers of tissues as per data availability. Broadly, we observe that active marks are significantly greater in MethExp-CGIs compared to both MethNotExp- (DNase: 10−14 < Wilcoxon P < 10−4 in 12/12 tissues; H3K4me3: 10−16 < Wilcoxon P < 10−4 in 32/32 tissues; H3K9ac: 10−17 < Wilcoxon P < 10−5 in 10/10 tissues) and NotMethExp-CGIs (DNase: 10−3 < Wilcoxon P < 0.05 in 8/12 tissues; H3K4me3: 10−3 < Wilcoxon P < 0.05 in 23/32 tissues; H3K9ac: 10−6 < Wilcoxon P < 0.05 in 8/10 tissues). In the case of repressive marks, while we broadly observe that they are significantly lower in MethExp-CGIs than MethNotExp- (H3K27me3: 10−9 < Wilcoxon P < 0.05 in 18/32 tissues; H3K9me3: 10−8 < Wilcoxon P < 0.05 in 30/32 tissues) and NotMethExp-CGIs (H3K27me3: 10−3 < Wilcoxon P < 0.05 in 18/32 tissues; H3K9me3: 10−3 < Wilcoxon P < 0.05 in 21/32 tissues), the differences in these levels are not as pronounced or widespread across tissues as for the active marks. This is consistent with the idea that active repression of an orphan CGI when the locus is not acting as an alternative promoter probably occurs less often and that these, in general, tend to prevail as open, accessible actively transcribing entities across the genome.
Evidence of transcriptional elongation and splicing occurring between distal CGIs and their associated MethExp gene promoters
The emerging trend of strong transcription initiation at distal CGIs that are associated with the expression of downstream MethExp genes, accompanied by a total lack of transcription initiation at proximal promoters, motivated us to probe further for evidence of bona fide promoter action at the CGIs, which we describe in four complimentary analyses that follow. It is known that any transcriptional activity ensuing from intergenic regulatory elements (i.e., true orphan CGIs and enhancers) that are not immediately proximal to coding or noncoding RNA genes does not culminate in the production of long RNA molecules (De Santa et al. 2010; Andersson et al. 2014). However, if indeed MethExp-associated CGIs function as promoters, they are expected to exhibit sustained transcriptional activity and elongation along the entire stretch of the intervening genomic region between the CGI and the downstream gene (henceforth referred to as the “segment”). While the presence of coding or noncoding genic elements in the segment region can bias quantitative measures of elongation, as mentioned earlier, this complication was preempted by excluding any such genes from our analyses (see Methods).
To this effect, first, we binned the segment region corresponding to MethExp, MethNotExp, and NotMethExp genes into 10 equal-sized bins and quantified in each bin three parameters that inform the extent of transcriptional activity as well as elongation: (1) RNA-seq signal strength (RPKM); (2) RNA-seq signal coverage (fraction of nucleotides supported by a read); and (3) H3K36me3 ChIP-seq signal (histone mark associated with the gene bodies of actively transcribed genes) (Hon et al. 2009). As can be seen in Figure 3, A–C, the segment region corresponding to MethExp genes shows significantly greater evidence for transcriptional activity and elongation than those for MethNotExp and NotMethExp genes (10−102 < Wilcoxon P < 10−5) in all tissues (with the exception of H3K36me3, wherein 31/33 and 29/33 tissues show significant trends, respectively). We present in the main text only the pooled distribution across all segment bins for each of the above, but the trends remain qualitatively similar in each assessed bin (Supplemental Fig. 9). These results are consistent with our prediction that CGIs associated with MethExp genes have a greater tendency to produce long RNA molecules extending into the body of the downstream gene.
Figure 3.

Transcriptional elongation and splicing signals between CGI and the gene. (A) Median RNA-seq RPKM signal, (B) median RNA-seq coverage, and (C) median H3K36me3 ChIP-seq signal and 95% CI (x-axes) associated with the segment region of MethExp (yellow), MethNotExp (red), and NotMethExp (green) genes across 34 tissue types (y-axes). (D) The average number of paired-end RNA-seq reads (y-axis) whose ends lie in both the segment region as well as the annotated gene, as seen across MethExp (yellow), MethNotExp (red), and NotMethExp (green) genes across all tissue types (x-axis). (E) The proportion of “stable” (orange) vs. “unstable” (dark blue) transcripts (y-axis), as determined from a sequence-based predictor (U1-PAS motif order in segment region) in those genes that are MethExp in at least one tissue, versus other genes (Fisher's P = 10−5). (F) Odds ratio and 95% CI (y-axis) of the proportion of (1) MethExp versus MethNotExp genes (cyan), and (2) MethExp versus NotMethExp genes (pink) that show evidence of splice junctions between the segment region and annotated gene based on de novo transcript assembly across 28 tissue types (x-axis). An enrichment of such splice junctions in the segment region associated with MethExp genes corresponds to a higher odds ratio. Statistically significant associations (P < 0.05) are annotated with an asterisk in all plots, in a matched color scheme, wherever appropriate. In panels A through C, this color corresponds to the color of the background gene group that MethExp genes are contrasted against, i.e., a red asterisk to represent significant difference between MethExp and MethNotExp, and green for that against the NotMethExp group.
As Pol II-Ser2 is also a marker of transcriptional elongation, we analyzed the Pol II-Ser2 signal in the segment region of MethExp, MethNotExp, and NotMethExp genes in MCF-7 cells (Supplemental Fig. 10). While the effect size of the trend (greater elongation in MethExp-segment regions compared with other groups) using Pol II-Ser2 is not as strong as when using RNA-seq and H3K36me3 data, MethExp-segments do show significantly greater elongation signals compared to MethNotExp (Wilcoxon P < 10−3).
Second, paired-end (PE) RNA-seq reads whose pairs are split across the segment and the downstream annotated gene region would provide a more direct indication for transcription initiating at upstream CGIs and extending into the body of MethExp genes. Such reads are not expected in the case of MethNotExp and NotMethExp genes because, in both cases, transcription initiates at the annotated primary promoter of these genes and not their associated upstream CGI. As expected, the proposed evidence is much greater for MethExp genes relative to the other two classes (Wilcoxon P < 10−4 in both cases) (Fig. 3D).
Third, it has been shown that transcripts that initiate from intergenic regulatory elements as well as those that remain unspliced terminate prematurely and are rapidly cleared away from the cell due to their instability in the absence of splice signals (Almada et al. 2013; Ntini et al. 2013). Previous studies have shown that sequence motifs dictate the production of stable vs. unstable transcripts; presence of a splice donor site facilitates binding of splicing factor U1 which can suppress polyadenylation site (PAS)-dependent termination, thereby promoting elongation of mRNAs (recently shown to be true in all transcript classes) (Schwalb et al. 2016). Core et al. (2014) used this in a hidden Markov model (HMM) and showed that U1 binding sites strongly tend to precede PAS on stable transcripts but not on unstable transcripts. Thus, we directly probed the order of occurrence of the above motifs in the sequence of the segment region to inform the stability of transcripts originating from the upstream CGIs associated with all genes. Each gene was deemed “stable” or “unstable” based on the order of the two motifs from the 5′ end of the segment. We then compared the fraction of stable transcripts between genes that are MethExp in at least one tissue to the rest of the genes using Fisher's exact test. We found that the fraction of “stable” transcripts is significantly greater among MethExp genes than among other genes (Fisher's P = 10−5) (Fig. 3E); however, the effect size is modest (Odds ratio = 1.2).
Finally, while it is not unrealistic for the region intervening between the distal CGI and the gene TSS to possess long 5′ UTRs (untranslated regions, which in eukaryotes can be up to several kb long) (Lodish 2008), it is more likely that it is spliced out in the mature transcript. Therefore, we directly assessed splicing activity by assembling transcripts de novo from RNA-seq reads using STAR (Dobin et al. 2013) and mapping splice junctions (see Methods). From the mapped junctions in each of 28 tissues (limited by raw read data availability), we quantified the number of MethExp, MethNotExp, and NotMethExp genes that showed evidence of a splice junction connecting their associated CGIs to their coding region (henceforth called a “split junction”). We found strong support for enrichment of split junctions in MethExp genes compared to both MethNotExp (8 < Odds Ratio < 340; 0 < Fisher's P < 10−67) and NotMethExp (2 < Odds Ratio < 23; 10−5 < Fisher's P < 10−40) genes in all 28 tissues (Fig. 3F). We illustrate one such example of GIGYF1, which is a MethExp gene in esophagus tissue (Fig. 4). Alternative promoter activity of its associated upstream unmethylated CGI is apparent in this case, where ensuing transcripts have their segment-region–spanning intron spliced out.
Figure 4.

An illustrative example. Transcriptomic and epigenetic marks surrounding a gene (GIGFY1) that is expressed despite a hypermethylated promoter in esophagus tissue. As shown, the proximal promoter is highly methylated (red corresponds to high methylation, whereas green corresponds to low), and yet there is a large signal for gene expression, as can be seen in the RNA-seq signal track. An upstream CGI (in pink) >6 kb away is free of methylation, and transcription of the gene ensues at this locus extending into the body of the gene. These patterns suggest that (1) the longest transcript starts at the CGI as opposed to its annotated start site in Ensembl/GENCODE, (2) the first intron spans the segment region and extends into the body of the gene (in cyan), and (3) there is a large splice junction (loop in dark red) that is split between a region located inside the segment and an exon inside the annotated gene.
Taken together, these results strongly suggest that CGIs associated with MethExp genes are bona fide promoters that produce transcripts that are stably elongated and spliced into the annotated genes.
Aberrant gene expression in cancer linked to hypomethylated distal CGIs
The aberrant DNA methylation landscape associated with cancer cells is considered to be a hallmark of the disease. Cancer is characterized by both global hypomethylation, as well as widespread promoter-associated hypermethylation of important genes like tumor suppressors (Jones and Baylin 2007), that lead to their silencing. We aimed to investigate the extent to which the usage of upstream CGIs as alternative promoters explains the aberrant gene expression patterns observed in cancer phenotypes.
We obtained RNA-seq and Illumina methylation array (450K) data from The Cancer Genomic Atlas (TCGA) for 780 breast cancer (Koboldt et al. 2012) and 315 renal cell carcinoma (Creighton et al. 2013) patients. Due to low coverage of 450K methylation data, only ∼3030 genes could be used for which methylation at both upstream CGI and promoter were available. Overall, there exist ∼300 (∼10%) methylated-promoter genes in each cancer sample that are expressed at high levels (see Methods). Similar to normal cells, these are also mainly protein-coding genes (85%) with mostly non-CGI promoters (90%). Given that hypermethylation in cancer is mainly targeted to CGI-promoters of genes (Sproul et al. 2012), one might expect to see a greater fraction of CGI-promoter genes in the MethExp group, but this was not the case. This strongly supports the idea that methylation at CGI-promoters is almost always accompanied by systematic silencing of that gene locus.
We find that CGIs associated with MethExp genes in cancer cells exhibit very similar properties to those found in normal tissues. Relative to MethNotExp-associated CGIs, MethExp-associated CGIs (1) have significantly lower methylation, (2) are closer to their associated gene loci, and (3) show significantly higher transcriptional activity and elongation (based on RNA-seq RPKM and read coverage measures) signals in the segment region. Figure 5 shows these trends for 100 representative breast and kidney cancer samples. As seen, the two gene groups are significantly different in all the above aspects (10−60 < Wilcoxon P < 10−8 across all comparisons). Thus, the use of distal CGIs by non-CGI methylated-promoter genes as alternative promoters is a general phenomenon, observed in both normal and cancer cells.
Figure 5.

Use of a distal CGI as an alternative promoter by MethExp genes in cancer. The figure shows four lines of evidence supporting the usage of a distal CGI as alternative promoter by MethExp genes in contrast to MethNotExp genes in (A) breast and (B) kidney cancer. Each panel shows the distribution of the median (1) fractional methylation at upstream CGIs (top left), (2) genomic distance between distal CGI and gene (bottom left), (3) RNA-seq RPKM signal (top right), and (4) RNA-seq coverage (bottom right) at the segment region (y-axes) corresponding to MethExp (yellow) vs. MethNotExp genes (red) across 100 representative samples (x-axes).
Next, we mapped the functional landscape of MethExp genes in cancer. First, we focused specifically on those sets of genes whose promoters are hypermethylated in cancer and that potentially rely on a distal CGI to express themselves. To this end, in a given cancer type, we identified genes whose promoters are hypermethylated in cancer (in >75% of the samples) and whose expression is associated with CGI methylation (Spearman's ranked correlation P < 0.05) but not with proximal promoter methylation (Spearman's P > 0.05). This resulted in 34 genes in breast and 39 genes in kidney cancer (listed in Supplemental Table 4). GO terms associated with general phenotypes in cancer like cell growth, maintenance or adhesion (P < 0.05) are overrepresented in both cases. More interestingly, we found an enrichment of protein sequence features and domains (FDR < 0.05) associated with (1) EGF, EGF-like, and palmitate among genes identified in breast (involved in breast cancer drug resistance) (Fig. 6A; Liu et al. 2008; Masuda et al. 2012), and (2) calcium binding, FOX transcription factor family, and alpha-actinins among genes identified in kidney cancer (key genes/pathways involved in decreased kidney function and cancer) (Fig. 6B; Linehan et al. 2010; Feng et al. 2015). This suggests that key genes involved in cancer also bypass their inactive promoters and utilize distal CGIs for their expression.
Figure 6.
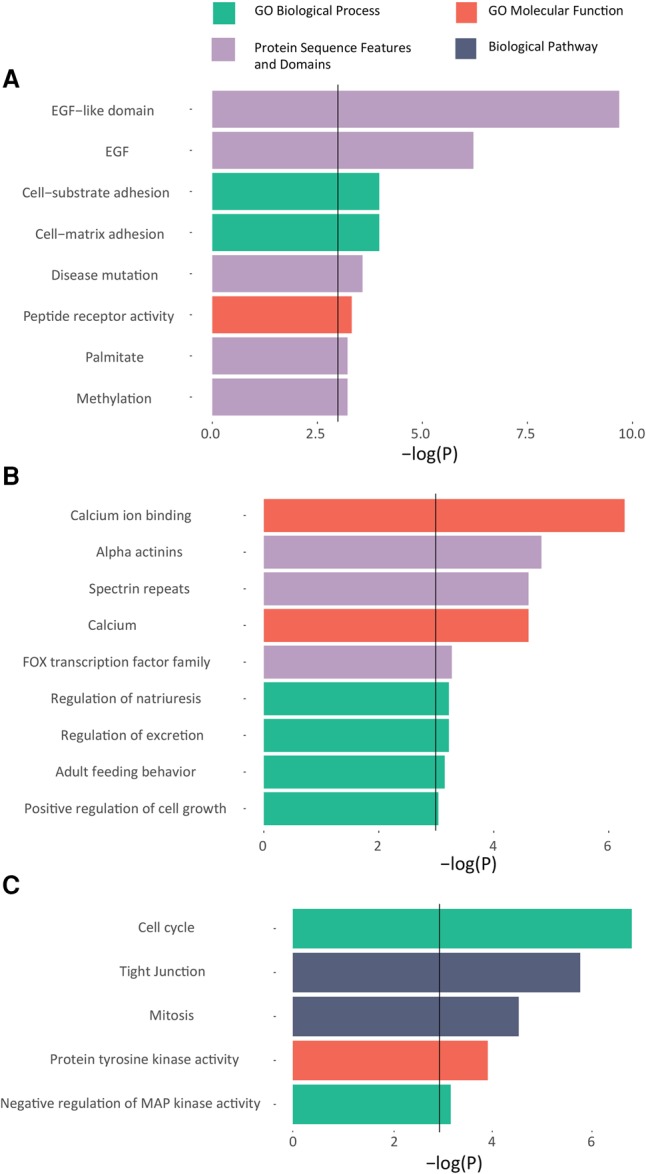
Functional enrichment of MethExp genes in cancer that potentially utilize an upstream CGI as a promoter. GO terms are shown on the y-axis, along with their corresponding −log(adjusted P) significance measures on the x-axis. Solid black line at P = 0.05 represents the threshold for enrichment. (A,B) Functional enrichment for genes whose promoters are broadly hypermethylated across samples but whose expression across samples is correlated with the upstream CGI's methylation and not that of the proximal promoter, in breast cancer (A), and (B) in kidney cancer. (C) Functional enrichment in genes whose differential expression between normal and breast cancer samples is correlated with the methylation status of the upstream CGIs and not with that of the proximal promoter.
Next, we focused specifically on genes that are differentially expressed in cancer relative to their normal counterparts, potentially due to differential methylation of their upstream CGI (and not the primary promoter). To this end, we obtained matched normal samples for 80 of the breast cancer samples (normal and cancer tissue from the same individuals) and identified 208 genes that are differentially expressed between cancer and normal samples (Wilcoxon P < 0.05) and whose nonzero expression is associated with CGI methylation (Spearman's P < 0.05) but not with proximal promoter methylation (Spearman's P > 0.05). These genes are enriched (FDR < 0.05) for GO terms related to cell cycle, cell growth (tyrosine and MAPK kinase signaling), and cell-cell adhesion, which are implicated in cancer progression and metastasis (Fig. 6C). Many of these genes exhibit very high negative correlation between upstream CGI methylation status and gene expression across healthy and cancer samples (up to Spearman's rho = −0.45). These include the YES1 (YES proto-oncogene 1, Src family tyrosine kinase), whose paralog LYN is involved in mediating treatment resistance in breast cancer (Schwarz et al. 2014), and the GINS2 gene, whose protein product interacts with CHEK2, a tumor suppressor gene linked to many cancers, including breast (Rantala et al. 2010). An entire list of these genes with their functional profiles is provided in Supplemental Table 5.
In summary, this previously unreported phenomenon, whereby distal CGIs are utilized as alternative promoters by certain highly expressed genes with methylated proximal promoters, is prevalent across several clinically important genes in cancer and warrants further investigation to chart its full implications.
Discussion
CpG islands were first discovered in mouse DNA in the '80s, in seminal work by Adrian Bird and others (Bird et al. 1985). Their unusually high frequency of CG dinucleotides (which are primary targets of DNA methylation in vertebrates), their virtually free-of-methylation disposition (in an otherwise globally methylated genome), as well as the fact that they surround the control regions of most genes led to their quick recognition as important regulatory elements. As a consequence, many of the studies that followed focused mainly on promoter proximal CGIs, which, incidentally, also happen to inform much of our understanding of the role of methylation in controlling chromatin structure and gene expression across tissues (Jones 2012). However, it was found that promoter-distal CGIs, despite being remote from annotated TSSs, were also capable of transcription initiation (promoter function) (Maunakea et al. 2010), and some of these sites were implicated in transcribing alternative tissue-specific isoforms (Hoivik et al. 2013) as intragenic alternative promoters or noncoding transcripts involved in imprinting and other functions (Mancini-DiNardo et al. 2003). Furthermore, it is the promoter-distal CGIs (orphan CGIs) that are more often differentially methylated, compared to promoter CGIs (Eckhardt et al. 2006), implicating them in condition-specific regulation. Despite these critical observations about orphan CGIs, a global view of their functional significance is only just beginning to emerge.
Here, we report a previously unknown phenomenon, whereby an intergenic orphan CGI can function as an alternative promoter to express the gene product of a nearby CpG-poor methylated-promoter gene. We found this to occur across hundreds of CpG-poor promoter genes that become methylated in a tissue-specific fashion. In an effort to assess the prevalence of alternative promoter usage of CGIs among the pool of MethExp genes, we quantified the broad features suggestive of alternative promoter usage, such as CGI methylation, CGI CAGE, and RNA Pol II-Ser5 (latter only in MCF-7), as well as segment RNA-seq signal and coverage, and computed the percentage of MethExp loci per tissue type that showed strong evidence of these based on stringent thresholds (see Supplemental Table 6 for details). As shown, the fractions of loci with strong support for alterative promoter use are quite high for all of the above features across cell types. This suggests that the usage of upstream CGIs as alternative promoters by genes with silenced proximal promoters is widespread.
Further, from the perspective of all orphan CGIs upstream of a CpG-poor promoter gene (within 50 kb), we find that almost 15% of them exhibit significant correlation (Spearman's P < 0.05) between CGI methylation and gene expression and lack such a correlation (Spearman's P > 0.05) between the gene's proximal promoter methylation and expression. Further, among all orphan CGIs exhibiting the above property, the corresponding downstream genes are significantly enriched for CpG-poor promoters (Fisher's P < 10−3) compared to CGI promoters. Even more interestingly, we find that the predominantly CpG-poor (i.e., non-CGI) promoters of MethExp genes tend to be more CG-rich than the average non-CGI promoter gene (Wilcoxon P = 10−9). It is possible that CpG-poor MethExp promoters are remnants of once CpG-rich promoters that have lost CG dinucleotides (due to the mutagenic property of methylcytosines) over evolutionary time; the overall impact of this phenomenon, however, remains unclear.
While the link between CGIs and downstream gene expression can be construed as a mode of distal enhancer-mediated regulation instead of alternative promoter action, we did not find any support to sustain that notion. We found no enrichment of tissue-specific ChromHMM annotated enhancers in MethExp CGIs across 30 tissues (Fisher's P = 0.4), and this is consistent with the established knowledge that enhancers are typically CpG-poor and are depleted of CGIs (Illingworth et al. 2010; Kim et al. 2010). Further, in addition to observing an enrichment of splice junctions between CGIs and their corresponding MethExp genes, we find some evidence of sequence-based predictors that support long, elongating, stable directional transcript production from MethExp-associated CGIs. These findings are in conflict with an enhancer model, as it is well known that any transcriptional activity at active enhancers results in short, typically unstable, bidirectional RNA (eRNAs) (De Santa et al. 2010; Kim et al. 2010).
Our findings caution against relying exclusively on proximal TSS platforms in determining the transcriptional outcome of a gene and implores us to extend focus to alternative distal elements, especially upstream orphan CGIs as they possess a “promoter-like” configuration. Very recently, a study that mapped the processes underlying the evolution of stripped-down retrocopies (intronless and promoterless copies of reverse-transcribed RNA inserted into the genome) into new bona fide functional genes discovered that only a marginal fraction (∼11%) of these retrocopies piggybacked on existing promoters for their expression, while the majority (∼86%) co-opted orphan CGIs and other proto-promoter elements (Carelli et al. 2016). Furthermore, as retrocopies emerged into fully functional genes, most (75%–93%) gained new exons from their upstream flanking sequences, and this overrepresentation of novel 5′ exons suggests that such a gain served to place them under the control of distal promoters, including orphan CGIs.
Nevertheless, the specific molecular mechanisms underlying the context-specific choice of proximal versus distal promoter in the case of MethExp genes remain unclear. While we cannot exclude the possibility that through some hitherto unknown mechanism, the usage of CGI is actively influenced by the methylation status of the proximal promoter, it is also possible that use of the alternative CGI promoter leads to transcriptional silencing of the proximal promoter, consistent with known patterns of high gene body methylation at highly transcribed regions (Laurent et al. 2010). However, our data and the results generally suggest that the usage of CGI occurs independent of the methylation status of the proximal promoter. First, the overall ability of distal CGIs to initiate transcription (evidenced by CAGE tags, for instance) seems largely independent of the methylation status of the proximal promoter (Fig. 2B,D,F). Second, while active histone marks are consistently much higher at MethExp-CGIs than NotMethExp-CGIs (i.e., at loci that are actually used as alternative promoters versus those that are not), the difference in the levels of repressive marks between these groups is not as pronounced or consistent across tissue types (Supplemental Fig. 8), suggesting that active repression of upstream CGIs occurs (if it does) independent of the methylation status of the proximal promoter. Thus, it most likely appears that a MethExp gene utilizes (or co-opts) an already active orphan CGI as an alternative promoter, analogous to the co-option of CGIs as promoters by promoter-less retrocopies of genes discussed above.
It seems likely that MethExp-associated CGIs have been co-opted relatively recently (at a time close to the divergence of mammals from the vertebrates analyzed in this study) for their regulatory role as alternative promoters. First, gene promoters that are more susceptible to silencing by methylation (namely, CpG-poor promoters) are associated more often with alternative promoter CGIs than CpG-rich promoters and appear to “co-opt” their usage in specific contexts (as evidenced by locus-specific CAGE analyses). Second, methylated-promoter genes and their upstream CGI elements are more likely to have conserved synteny when they are expressed, and importantly, this tendency increases monotonically as more closely related species are used to ascertain the synteny, suggesting an evolutionary selection to keep the segment region intact. Finally, orphan CGIs have been shown to be co-opted by promoter-less genes in humans (i.e., retrocopies) to transcribe their gene products which, together with our findings, suggests that this is a general property of orphan CGIs. Thus, a more holistic view of the biological significance of CGIs is beginning to emerge in that they are ubiquitous substrates that are poised as transcriptional initiation sites that, in a contextually favorable configuration (i.e., unmethylated and upstream of a stable RNA producing transcription elongation–enhancing element), can be selected for alternative promoter activity by a proximally located neighboring gene.
Methods
Data sets
Expression
RNA-seq expression for 30 primary tissues and four ENCODE cell lines analyzed in this study were obtained from Release 9 of the compendium published by the NIH Roadmap Epigenomics Project (Bernstein et al. 2010). This release comprised uniformly reprocessed data for 111 consolidated epigenomes (111 primary tissue types) (Kundaje et al. 2015), wherein each sample from their original source underwent additional processing in an effort to reduce redundancy, improve quality control, and achieve uniformity for integrative analysis. Raw read and processed data are publicly available and were both used in this study.
Methylation
We limited our analyses to tissues with publicly available WGBS data that were also sourced from the consolidated epigenomes work. Methylation measures for every CpG dinucleotide were provided in the format of fractional methylation (Reads recording a methylated CpG/Total Reads). BED files with read depth and fractional methylation information are publicly available.
Annotation
The specific version of hg19 genome annotation used in the consolidated epigenomes work cited above was GENCODE v10 (corresponding to Ensembl v65) (Harrow et al. 2012) and has therefore been carried forward in all the analyses performed in this study to maintain consistency. To verify that our results were robust with respect to the latest assembly of the human genome (GRCh38), we repeated a few key analyses in one cell type using the GRCh38 gene annotations and the “lifted-over” RNA-seq and methylation data. We observed that the overall trends for differences in CGI methylation levels, genomic distance, and transcriptional elongation signals between upstream CGI and gene between the MethExp vs. the MethNotExp categories are consistent between the two versions (Supplemental Fig. 11).
CpG islands
Annotations of CpG islands were extracted from the UCSC Genome Browser. This track corresponds to a hierarchical HMM model-based definition of CpG islands in hg19 (Irizarry et al. 2009b; Wu et al. 2010).
Syntenic blocks
Precomputed syntenic blocks derived from whole-genome sequence alignments between human (as the reference) and six mammalian species (chimpanzee, rhesus monkey, mouse, rat, dog, and cow) as well as two nonmammalian vertebrate species (chicken and zebrafish) were downloaded from CINTENY (Sinha and Meller 2007).
CAGE
Single-molecule CAGE profiles for 573 human primary cell samples (up to a median depth of 4 million mapped tags per sample) were generated by the FANTOM Consortium (The FANTOM Consortium and the RIKEN PMI and CLST (DGT) 2014). Out of the 34 tissues we analyzed, CAGE was available for 15 of them. These data are preprocessed to report the CAGE peaks associated with TSSs found genome-wide and are available as BED files. To determine the CAGE tag level in a given genomic region (for example, promoters or CGIs), we used the dominant TSS or the TSS with the highest number of CAGE tags.
Ser5P and Ser2P RNA Pol-II ChIP-seq
ChIP-seq assays targeted to Ser5 and Ser2 phosphorylated molecules of RNA Pol-II are currently limited to only one cell type (MCF-7) used in our analyses. Fold-change signal data at base-pair resolution were obtained from GEO accession GSE54693 (Menafra et al. 2014).
Histone marks and DNase-seq
Processed data at base-pair resolution for several histone marks and DNase-seq (cleaving DNase hypersensitivity sites) are available from the consolidated epigenomes work cited above.
Data sets used in mouse DNMT knockout analysis
RNA-seq and WGBS methylation data for mouse wild-type and DNMT knockout embryonic stem cells were obtained from GEO accession number GSE67867 (Domcke et al. 2015). They mapped their data to the mm9 genome assembly version of mouse (NCBIM37) and made available read density and fractional methylation at base-pair resolution. Whole-genome sequence, CGI and gene annotation files corresponding to mm9 were downloaded from Ensembl and the UCSC Genome Browser.
Data sets used in cancer-related analyses
Data for 780 breast cancer samples, 80 matched breast normal samples (matched to their corresponding cancer samples from the same individuals), and 315 renal (kidney) cell cancer samples from TCGA (Koboldt et al. 2012; Creighton et al. 2013) were downloaded using the CGHub Repository (Wilks et al. 2014). Data for each sample comprised 450K methylation arrays (reporting fractional methylation at select CpG probes) and RNA-seq expression (raw read file FASTQ and processed gene expression in RPKM). To obtain measures of transcriptional activity in the “segment” region (RPKM and read coverage), raw reads from each sample were aligned using STAR (Dobin et al. 2013) and further processed using the BEDTools suite (Quinlan and Hall 2010).
Primary processing of genes and pooling into gene groups
The promoter of a gene was marked as methylated when the average fractional methylation level of all CpG dinucleotides lying within TSS ± 500 bp was greater than 0.55, and unmethylated when that value was less than 0.45. As vertebrate promoters exhibit a clear bimodal pattern of lowly and heavily methylated promoters (Elango and Yi 2008), we consider the above thresholds to be fairly stringent. Yet, to be certain that we indeed captured only the highly methylated class of promoters in our MethExp category of genes per cell type using the above threshold, we conducted the following sanity check. We fit a three-component Gaussian mixture model to the overall distribution of promoter methylation levels per tissue type to distinguish three subpopulations corresponding to lowly, intermediate, and highly methylated promoters (LMP, IMP, HMP), and then checked the fraction of MethExp promoters, selected based on the aforementioned threshold, belonging to HMP separately in each tissue type. We found that, on average, ∼97.6% of them belong to HMP (Supplemental Fig. 12). Further, a gene was considered “expressed” if its expression was in the top 50th percentile among all genes. The threshold adopted for expression is highly stringent and conservative since we wanted to focus on explaining the mechanisms adopted by highly expressed genes with methylated primary promoters. A gene was considered as not expressed when it had zero expression or its expression value was in the bottom 5th percentile among all genes. The above criteria were used to pool genes into three gene groups, MethExp, MethNotExp, and NotMethExp, in each sample.
The distal CGI associated with a given gene was defined as the closest upstream CGI annotated at a minimum distance of 1500 bp from TSS. Most annotated CGIs are <1 kb long (∼83%). Those longer than 1 kb were truncated to centerpoint ± 500 bp for the computation of methylation levels. This did not affect the estimation of methylation levels, as these distributions are almost identical before and after CGI truncation (Supplemental Fig. 13). Additionally, we discarded from all three groups every gene that contained another annotated gene between its TSS and upstream CGI element. This annotated gene could be an ambiguous ORF or any noncoding RNA including lincRNAs, overlapping sense or antisense RNAs/genes, snRNA, tRNAs, etc., annotated by GENCODE. This was done to ensure that there existed no biases from neighboring genes on our observations of intergenic transcriptional activity or neighboring epigenetic and chromatin signatures.
Evidence of gene body alternative promoter usage
To identify the fraction of MethExp genes that initiate transcription from a locus within the gene body distinct from its proximal promoter, we quantified the expression level of all exons within each gene. Then, for each MethExp gene, if the expression level (RPKM) of the first exon was zero or in the bottom 5th percentile among all exons of all genes, then that gene was concluded to possess a silenced primary promoter with an active gene body alternative promoter.
Tissue specificity index (TSI)
The quantitative measure of TSI, is defined as
where N is the number of tissues and xi is the expression profile component normalized by the maximal component value (Yanai et al. 2005).
Evolutionary conservation
Conservation of distal CpG islands was calculated at two distinct levels.
Interspecies conservation
We used genome-wide base-pair resolution phastCons scores that were precomputed from the multiple sequence alignment of 45 vertebrate genomes to the human genome (Siepel et al. 2005).
Intraspecies conservation
We used genome-wide human polymorphism data from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012) to infer the extent of intraspecific selection pressure acting on distal CGI elements. A derived allele is one that arises in a population due to a mutation in the original allele in the population (ascertained by comparing with multiple closely related species to human). By definition, the derived allele starts out “rare,” and its frequency can increase in a population over time due to genetic drift or, on rare occasions, positive selection. If the mutation or the derived allele is deleterious, its spread will be curtailed due to selection pressures acting on it, thereby resulting in a low derived allele frequency (Vishnoi et al. 2011). Therefore, a low DAF in a given region may suggest negative selection in that region. For each CGI, we generated the DAF spectrum by pooling DAFs at all nucleotides within that region. Thus, for each gene locus, there existed a DAF profile corresponding to its upstream CGI.
Cell type-specific regulation of alternative promoter CGIs
Motif information for 642 TFs (those with available Positional Weight Matrix [PWM] in TRANSFAC [Matys et al. 2006] and expression data across cell types) and the sequences of all CGIs showing evidence of alternative promoter activity in some cell type were input to PWMSCAN (Levy and Hannenhalli 2002), a tool that scans sequences to identify significant motif matches. Matches with PWM scores in the top 5% were retained, and expression profiles of the corresponding TF genes were obtained. Then, for each locus, the distribution of the expression profiles of these TFs in cell types where the CGI was active was compared to a similar distribution arising from cell types where the CGI was inactive, using the Wilcoxon test.
Sequence-based splicing signals
Sequences spanning the intergenic region between the TSS of MethExp, MethNotExp, and NotMethExp genes and their associated upstream distal CGIs (“segment region”) were extracted using the hg19/GRCh37 reference genome from the UCSC Genome Browser. Motif information and frequency matrices for the U1 binding site and PAS recognition sequence were obtained from Almada et al. (2013). The motif frequency data were transformed to position weight matrices and was input to PWMSCAN (Levy and Hannenhalli 2002), a tool that scans sequences to identify significant motif matches. Matches with PWM scores in the top 5% were retained, and the order of motifs on a given sequence was inferred. If the first 1500 bp of the segment region contained a match for U1 before PAS, the corresponding gene locus was assigned the label “stable” and “unstable” in case the motif order was switched.
Gene Ontology (GO) enrichment
DAVID Bioinformatics Resource 6.7 (Dennis et al. 2003) was used for all GO enrichment and functional annotation performed in this study.
Supplementary Material
Acknowledgments
We thank Steve Mount and Soojin Yi for their useful comments and suggestions. This work was supported by NIH grant R01GM100335 to S.H.
Author contributions: C.V. conceived the project. S.H., S.S., and A.D. designed the study. S.S. carried out the analysis, and S.S. and S.H. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.212050.116.
References
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. 2013. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499: 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Arthur L, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. 1985. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40: 91–99. [DOI] [PubMed] [Google Scholar]
- Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, Temper V, Razin A, Cedar H. 1994. Sp1 elements protect a CpG island from de novo methylation. Nature 371: 435–438. [DOI] [PubMed] [Google Scholar]
- Carelli FN, Hayakawa T, Go Y, Imai H, Warnefors M, Kaessmann H. 2016. The life history of retrocopies illuminates the evolution of new mammalian genes. Genome Res 26: 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. 2014. Analysis of transcription start sites from nascent RNA supports a unified architecture of mammalian promoters and enhancers. Nat Genet 46: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Gordon Robertson A, Chu A, Beroukhim R, Cibulskis K, Signoretti S, et al. 2013. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei C-L, Natoli G. 2010. A large fraction of extragenic RNA Pol II transcription sites overlap enhancers. PLoS Biol 8: e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton A, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Webb S, Kerr ARW, Illingworth RS, Guy J, Andrews R, Bird A. 2011. Cell type–specific DNA methylation at intragenic CpG islands in the immune system. Genome Res 21: 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3. [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D. 2015. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528: 575–579. [DOI] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. 2006. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38: 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango N, Yi SV. 2008. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol Biol Evol 25: 1602–1608. [DOI] [PubMed] [Google Scholar]
- The FANTOM Consortium and the RIKEN PMI and CLST (DGT). 2014. A promoter-level mammalian expression atlas. Nature 507: 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, DuMontier C, Pollak MR. 2015. The role of α-actinin-4 in human kidney disease. Cell Biosci 5: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaumet-Adkins A, Richter J, Odero MD, Sandoval J, Agirre X, Catala A, Esteller M, Prósper F, Calasanz M, Buño I, et al. 2014. Hypermethylation of the alternative AWT1 promoter in hematological malignancies is a highly specific marker for acute myeloid leukemias despite high expression levels. J Hematol Oncol 7: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. 2011. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet 20: 4299–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoivik EA, Witsoe SL, Bergheim IR, Xu Y, Jakobsson I, Tengholm A, Doskeland SO, Bakke M. 2013. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One 8: e67925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Ren B. 2009. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet 18: R195–R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP. 2009. CpG islands—“a rough guide”. FEBS Lett 583: 1713–1720. [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr ARW, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. 2010. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet 6: e1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. 2009a. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Wu H, Feinberg AP. 2009b. A species-generalized probabilistic model-based definition of CpG islands. Mamm Genome 20: 674–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. 2007. The epigenomics of cancer. Cell 128: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. 2015. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Hemberg M, Gray J, Costa A. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, et al. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen F, Gundersen G, Lopez R, Prydz H. 1992. CpG islands as gene markers in the human genome. Genomics 13: 1095–1107. [DOI] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. 2010. Dynamic changes in the human methylome during differentiation. Genome Res 20: 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay FD, Liu Y, Kelly TK, Witt H, Farnham PJ, Jones PA, Berman BP. 2015. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res 25: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Hannenhalli S. 2002. Identification of transcription factor binding sites in the human genome sequence. Mamm Genome 13: 510–514. [DOI] [PubMed] [Google Scholar]
- Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schübeler D. 2011. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet 43: 1091–1097. [DOI] [PubMed] [Google Scholar]
- Linehan W, Srinivasan R, Schmidt L. 2010. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 7: 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu Y, Zhang J-T. 2008. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther 7: 263–270. [DOI] [PubMed] [Google Scholar]
- Lodish H, Berk A, Kaiser CA, Krieger M, Scott MP, Bretscher A, Ploegh H, Matsudaira P. 2008. Molecular cell biology. 6th ed W.H. Freeman, New York. [Google Scholar]
- Mancini-DiNardo D, Steele SJS, Ingram RS, Tilghman SM. 2003. A differentially methylated region within the gene Kcnq1 functions as an imprinted promoter and silencer. Hum Mol Genet 12: 283–294. [DOI] [PubMed] [Google Scholar]
- Martino D, Saffery R. 2015. Characteristics of DNA methylation and gene expression in regulatory features on the Infinium 450k Beadchip. bioRxiv 10.1101/032862. [DOI] [Google Scholar]
- Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. 2012. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat 136: 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menafra R, Brinkman AB, Matarese F, Franci G, Bartels SJJ, Nguyen L, Shimbo T, Wade PA, Hubner NC, Stunnenberg HG. 2014. Genome-wide binding of MBD2 reveals strong preference for highly methylated loci. PLoS One 9: e99603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, Yi SV. 2015. Whole-genome bisulfite sequencing maps from multiple human tissues reveal novel CpG islands associated with tissue-specific regulation. Hum Mol Genet 25: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moarii M, Boeva V, Vert J-P, Reyal F. 2015. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genomics 16: 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Zhang B, Bell RJA, Johnson BE, Olshen AB, Sundaram V, Li D, Graham AE, Diaz A, Fouse SD, et al. 2014. Recurrent epimutations activate gene body promoters in primary glioblastoma. Genome Res 24: 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntini E, Järvelin AI, Bornholdt J, Chen Y, Boyd M, Jørgensen M, Andersson R, Hoof I, Schein A, Andersen PR, et al. 2013. Polyadenylation site–induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol 20: 923–928. [DOI] [PubMed] [Google Scholar]
- Phatnani HP, Greenleaf AL. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 20: 2922–2936. [DOI] [PubMed] [Google Scholar]
- Pollard SM, Stricker SH, Beck S. 2009. A shore sign of reprogramming. Cell Stem Cell 5: 571–572. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantala JK, Edgren H, Lehtinen L, Wolf M, Kleivi K, Vollan HKM, Aaltola A-R, Laasola P, Kilpinen S, Saviranta P, et al. 2010. Integrative functional genomics analysis of sustained polyploidy phenotypes in breast cancer cells identifies an oncogenic profile for GINS2. Neoplasia 12: 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxonov S, Berg P, Brutlag DL. 2006. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci 103: 1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Fruhauf K, Demel C, Tresch A, Gagneur J, Cramer P. 2016. TT-seq maps the human transient transcriptome. Science 352: 1225–1228. [DOI] [PubMed] [Google Scholar]
- Schwarz LJ, Fox EM, Balko JM, Garrett JT, Kuba MG, Estrada MV, González-Angulo AM, Mills GB, Red-Brewer M, Mayer IA, et al. 2014. LYN-activating mutations mediate antiestrogen resistance in estrogen receptor–positive breast cancer. J Clin Invest 124: 5490–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilpa V, Bhagat R, Premalata CS, Pallavi VR, Ramesh G, Krishnamoorthy L. 2014. Relationship between promoter methylation & tissue expression of MGMT gene in ovarian cancer. Indian J Med Res 140: 616–623. [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha AU, Meller J. 2007. Cinteny: flexible analysis and visualization of synteny and genome rearrangements in multiple organisms. BMC Bioinformatics 8: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z, Chan M, Mikkelsen T, Gu H. 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproul D, Nestor C, Culley J, Dickson JH, Dixon JM, Harrison DJ, Meehan RR, Sims AH, Ramsahoye BH. 2011. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc Natl Acad Sci 108: 4364–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproul D, Kitchen RR, Nestor CE, Dixon JM, Sims AH, Harrison DJ, Ramsahoye BH, Meehan RR. 2012. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol 13: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. [DOI] [PubMed] [Google Scholar]
- van Eijk KR, de Jong S, Boks MPM, Langeveld T, Colas F, Veldink JH, de Kovel CGF, Janson E, Strengman E, Langfelder P, et al. 2012. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics 13: 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlodrop IJH, Niessen HEC, Derks S, Baldewijns MMLL, Van Criekinge W, Herman JG, Van Engeland M. 2011. Analysis of promoter CpG island hypermethylation in cancer: location, location, location! Clin Cancer Res 17: 4225–4231. [DOI] [PubMed] [Google Scholar]
- Vishnoi A, Sethupathy P, Simola D, Plotkin JB, Hannenhalli S. 2011. Genome-wide survey of natural selection on functional, structural, and network properties of polymorphic sites in Saccharomyces paradoxus. Mol Biol Evol 28: 2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. 2014. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol 15: R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Oliver VF, Wang G, Zhu H, Zack DJ, Merbs SL, Qian J. 2015. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics 16: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks C, Cline MS, Weiler E, Diehkans M, Craft B, Martin C, Murphy D, Pierce H, Black J, Nelson D, et al. 2014. The Cancer Genomics Hub (CGHub): overcoming cancer through the power of torrential data. Database (Oxford) 2014: bau093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. 2010. Redefining CpG islands using hidden Markov models. Biostatistics 11: 499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21: 650–659. [DOI] [PubMed] [Google Scholar]
- Zhu J, He F, Hu S, Yu J. 2008. On the nature of human housekeeping genes. Trends Genet 24: 481–484. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT-Y, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. 2013. Charting a dynamic DNA methylation landscape of the human genome. Nature 500: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.