

Abstract
Triple negative breast cancer (TNBC) is a very aggressive subgroup of breast carcinoma, still lacking specific markers for an effective targeted therapy and with a poorer prognosis compared to other breast cancer subtypes.
In this study we investigated the possibility that TNBC cells contribute to the establishment of tumor vascular network by the process known as vasculogenic mimicry, through endothelial cell differentiation.
Vascular‐like functional properties of breast cancer cell lines were investigated in vitro by tube formation assay and in vivo by confocal microscopy, immunofluorescence or immunohistochemistry on frozen tumor sections. TNBCs express endothelial markers and acquire the ability to form vascular‐like channels in vitro and in vivo, both in xenograft models and in human specimens, generating blood lacunae surrounded by tumor cells. Notably this feature is significantly associated with reduced disease free survival. The impairment of the main pathways involved in vessel formation, by treatment with inhibitors (i.e. Sunitinib and Bevacizumab) or by siRNA‐mediating silencing, allowed the identification of PDGFRβ and FGFR2 as relevant players in this phenomenon. Inhibition of these tyrosine kinase receptors negatively affects vascular lacunae formation and significantly inhibits TNBC growth in vivo.
In summary, we demonstrated that TNBCs have the ability to form vascular‐like channels in vitro and to generate blood lacunae lined by tumor cells in vivo. Moreover, this feature is associated with poor outcome, probably contributing to the aggressiveness of this breast cancer subgroup. Finally, PDGFRβ and FGFR2‐mediated pathways, identified as relevant in mediating this characteristic, potentially represent valid targets for a specific therapy of this breast cancer subgroup.
Keywords: TNBC, Vasculogenic mimicry, PDGFR, FGFR
Highlights
TNBCs show capability to perform in vitro and in vivo Vasculogenic Mimicry (VM).
In TNBC the n of vascular lacunae is significantly higher and associated with DFS.
Tumor cells performing in vivo VM express endothelial and mesenchymal markers.
Sunitinib strongly inhibits VM and induces tumor regression in TNBC models.
SiRNA‐mediated silencing revealed a crucial role for PDGFRβ and FGFR2 in VM.
Abbreviations
- CK
Cytokeratin
- DFS
Disease Free Survival
- EGFR
Epidermal Growth Factor Receptor
- ER
Estrogen Receptor
- FFPE
Formalin-Fixed Paraffin-Embedded
- FGFR
Fibroblast Growth Factor Receptor
- HER2
Human Epidermal Growth Factor receptor 2
- HPFs
High-Power Fields
- PDGFRβ
Platelet-derived Growth Factor Receptor
- PR
Progesterone Receptor
- SRB
Sulforhodamine B
- TEM
Transmission Electron Microscopy
- TKI
Tyrosin Kinase Inhibitor
- TNBC
Triple Negative Breast Cancer
- VEGF
Vascular Endothelial Growth Factor
- VEGFR
Vascular Endothelial Growth Factor Receptor
- VM
Vasculogenic Mimicry
- WGA
Wheat Germ Agglutinin
1. Introduction
Triple negative breast cancers (TNBCs) lack the three most significant therapeutic markers for clinical management of breast cancer patients, Human Epidermal Growth Factor receptor 2 (HER2), Estrogen Receptor (ER) and Progesterone Receptor (PR) (Dawood et al., 2009; Irvin, Jr. and Carey, 2008), and their prognosis is globally poorer than the other breast carcinoma subgroups. Therefore, lacking specific markers for an effective targeted therapy, TNBCs still represent the most important challenge for clinical oncologists.
There is a significant molecular heterogeneity within TNBC, which could explain the unsatisfactory activity of new therapies utilized so far in clinical trials. According to the first study reporting the intrinsic subclassification of breast carcinomas, where gene expression profiles were able to further classify breast carcinomas in different subgroups, TNBCs mainly belong to basal‐like subtype (Perou et al., 2000). Successively, Cheang et al. (2008) categorized TNBCs lacking basal Cytokeratin (CK) 5/6 and Epidermal Growth Factor Receptor (EGFR) expression (5 negative tumors), which actually account for about 50% of TNBCs. More recently, molecular stratification of TNBCs primarily using DNA microarrays and patterns of gene expression has identified that TNBCs can be classified as basal‐like (50%), claudin‐low (30%) and luminal A, B and HER2 subtypes for the remaining 20% distinct tumor subtypes (Perou, 2011; Prat and Perou, 2011). Finally, a further gene profiling of more than 500 TNBCs classified them in 6 different subtypes: basal 1 and 2, mesenchymal‐like and mesenchymal stem cell‐like, immunomodulatory and androgen pathway enriched (Lehmann et al., 2011).
Despite differences in molecular features, metastasis‐free survival analyses indicated that all TNBCs share a similar poor prognosis among the different subtypes (Ménard, 2011), suggesting that aggressiveness of these tumors may account for common characteristics. Supporting this hypothesis, TNBCs are all enriched in expression of mesenchymal/stromal genes compared to luminal and HER2 breast cancer, feature that might improve the crosstalk between tumor and microenvironment required for neoplastic progression. Moreover, it has been recently suggested that aggressive tumors including TNBCs are able to organize vessel‐like networks through trans‐differentiative ability of cancer stem cells (Liu et al., 2012). This process, called Vasculogenic Mimicry (VM), is probably able to provide nutrients to support TNBCs, contributing to the disease progression with the acquisition of a more aggressive and invasive phenotype (Kirschmann et al., 2012).
In this study, we deeply investigated the capability of TNBCs to differentiate into endothelial cells and participate in tumor vascularization. TNBC cells acquire indeed the ability to form vascular‐like channels both in vitro, when seeded on Matrigel, and in vivo, generating blood lacunae lined by tumor cells. Moreover, we identified Platelet‐derived Growth Factor Receptor β (PDGFRβ) and Fibroblast Growth Factor Receptor (FGFR) 2 pathways as relevant in mediating this characteristic, potentially representing a valid target for a specific therapy of this breast cancer group.
2. Methods
2.1. Cell lines, reagents and treatments
All cell lines were purchased from ATCC (Rockville, MD, USA) and maintained as recommended. MDA‐MB‐231‐GFP cells were kindly provided by Dr. Colombo (Fondazione IRCCS Istituto Nazionale dei Tumori, Milan).
SU11248/Sunitinib malate (Sutent) (Pzifer, New York, USA) was resuspended in methylcellulose and used at 40 mg/kg/day for 14 days (in vivo) or suspended in DMSO and used at 6.25 μM (in vitro); Bevacizumab (Roche, Basel, Switzerland) was diluted in physiological buffer and used in vivo (4 mg/kg/twice a week for 4 weeks) and in vitro (50 μg/ml). TIMP‐2 (MMP2 inhibitor) (Chemicon International, Billerica, MA, USA) was used at 1 μg/ml, whereas Zometa (zoledronic acid, MMP9 inhibitor) (Novartis, Basel, Switzerland) 5 μM in in vitro experiments. Imatinib (Selleck Chemicals, USA) and pan‐FGFR Inhibitor (PD173074) were purchased from commercial suppliers, resuspended in DMSO and used at 10 μM (in vitro). All compounds were used at a non‐cytotoxic concentration (as evaluated by Sulforhodamine B (SRB) colorimetric assay).
Controls of each treatment are represented by cells treated with the same amount of vehicle.
2.2. Immunofluorescence analysis
When tumors reached a volume of about 150 mm3, mice were injected i.v. with fluorescein Tomato lectin (VectorLabs, Burlingame, CA, USA) or with Alexa Fluor 555 Wheat Germ Agglutinin (WGA) (Invitrogen Life Technologies, Carlsbad, CA) for 15 min and then animals were killed. Tumors were excised, fixed with paraformaldehyde 4% and frozen processed.
Cryostat sections were permeabilized using PBS 0.1% TritonX100 and incubated with a blocking 2% BSA solution, primary antibodies, anti‐EGFR rabbit (Acris Gmbh, Germany); nuclear DRAQ‐5 (Biostatus Limited, UK); biotinilated anti‐rabbit antibody, and finally detected with Alexa Fluor 546 Streptavidin (Invitrogen Molecular Probes, Carlsbad, CA). Slides were analyzed with a confocal microscopy (Microradiance 2000; Bio‐Rad Laboratories) equipped with Argon/Krypton (488 nm), HeNe (543 nm) and Red Laser Diode (638 nm) lasers. Confocal images (512 × 512 pixels) were obtained using a 60× oil immersion lens and analyzed using ImagePro 7.0 software. Reported images represent a single frame from 15 to 16 stacks (0.5 μm step). The pin‐hole diameter was regulated according to the value suggested by the acquisition software to obtain the maximum resolution power.
2.3. Patient samples
Breast tissue samples obtained from patients with invasive breast carcinoma were selected from the archives of the Human Pathology Section, University of Palermo and of the Pathology Dept. of Fondazione IRCCS Istituto Nazionale dei Tumori of Milan. 87 TNBC cases (of which 77 with available follow‐up), diagnosed between December 2007 and January 2012, according to immunohistochemical criteria (lack of ER, PR and HER2 expression), were selected for this study. Thirty specimen from luminal (20 cases) and HER2+ (10 cases) patients were selected as controls. All the TNBC cases could be considered as grade 3 (G3) according to the Nottingham combined histologic grade (Ellis and Elston, 2006). Non‐TNBC group comprised tumors with different histological features and degree of pleiomorphism (8 cases of poorly differentiated G3, 18 cases of moderately G2 and 4 cases of well differentiated G1 tumors). All of the procedures were in accordance with the Helsinki Declaration (World Medical Association, 2013). Concerning biospecimens, leftover material of samples collected during standard surgical and medical approaches at Fondazione IRCCS Istituto Nazionale dei Tumori of Milan are used for research. Samples included in this paper have been donated by the patients to the Institutional BioBank for research purposes, and aliquots have been attributed to this study after approval of Institutional Review Board and specific request to the Independent Ethical Committee.
2.4. Histomorphological and immunohistochemical analysis
Four‐micrometers‐thick sections of formalin‐fixed and paraffin embedded specimens were routinely‐stained with Hematoxylin & Eosin for histomorphological evaluation. The number of vascular lacunae, both in human and murine samples, was assessed by counting the absolute number of vascular structures lined by neoplastic cells out of 5 high‐power fields (HPFs, ×400 magnification) and then averaging the counts of the 5 fields. Similarly, in murine samples, the average number of apoptotic figures out of the 5 HPF, was also evaluated.
Immunohistochemistry was performed as previously reported (Korpal et al., 2011). After microwave antigen retrieval, the sections were incubated overnight with mouse anti‐human/mouse CD31 (dilution 1:50, Novocastra) primary antibodies and staining was revealed by polymer detection kit (Novocastra).
3,31‐Diaminobenzidine (DAB; brown signal) was used as a chromogenic substrate, and sections were counterstained with Hematoxylin. Negative control stainings were performed by using mouse, rabbit, or goat immune sera instead of the primary antibodies.
For double‐marker immunofluorescence on breast tissue, sections underwent two sequential rounds of single‐marker immunostaining, as previously reported (Korpal et al., 2011). The samples were incubated with mouse anti‐human AE1/AE3 pan‐CK (dilution 1:100, Novocastra) or mouse/anti‐human p53 (dilution 1:50, Novocastra), and, after Fc blocking, with mouse anti‐human CD31 or CD34 (Novocastra). The binding was revealed using Alexa Fluor 488‐conjugated and Alexa Fluor 568‐conjugated goat anti mouse antibodies (dilution 1:300, Invitrogen Life Technologies), respectively.
2.5. Electron microscopy
Tumors were immersion‐fixed in 3% buffered glutaraldehyde for 2 h, post‐fixed in 1% osmium tetroxide and processed for epoxy resin embedding and thin‐sectioned.
2.6. In vitro tube formation assay
2 × 104 cells were seeded in 96‐well plates pre‐coated with Matrigel (BD Biosciences) (35 μl/well, diluted 1:1 in medium without FBS) and incubated for 4 h at 37 °C. Tube formation was acquired using a Nikon inverted light microscope (40×), and complete loops quantified by a macro made with the Image‐Pro Plus 7.0 software.
2.7. Knock‐down of PDGFRβ, FGFR2, FGFR1, VEGFR1, VEGF and EGFR by siRNA transfection
siRNA for human PDGFRβ, FGFR2, FGFR1, Vascular Endothelial Growth Factor Receptor (VEGFR) 1, Vascular Endothelial Growth Factor (VEGF) and EGFR (ON‐TARGET plus SMART pool, Dharmacon, Colorado, USA) or control siRNA (On‐TARGETplus Non‐Targeting Pool) were trasfected (100 nM) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were harvested at 24–48 h post‐transfection.
2.8. SRB assay
5000 cells/well (MDA‐MB‐231, MDA‐MB‐468 and MCF‐7) were seeded in 96‐well plates, treated with Sunitinib at different concentrations and with Bevacizumab at different concentrations for 3 days, then processed as previously reported (Skehan et al., 1990).
2.9. Cell cycle
Cell cycle of 5 × 105 RNAi‐silenced MDA‐MB‐231 and MDA‐MB‐468 cells was evaluated as previously described (Di Leva et al., 2010). The data obtained were analyzed using ModFit software, 3.2 version (Verity Software House).
2.10. Xenograft models
Experimental protocols were approved by the Ethics Committee for Animal Experimentation of Fondazione IRCCS Istituto Nazionale dei tumori of Milan. Six‐week‐old female SCID mice (Charles River, Wilmington, MA, USA) were given mammary fat pad injections of 5 × 106 MDA‐MB‐231, MDA‐MB‐468 and MCF‐7 cells diluted 1:1 in matrigel: RPMI 10% FBS (BD Biosciences, Canada). Mice inoculated with estrogen‐sensitive MCF‐7 cells were treated intramuscularly with weekly estradiol (0.12 mg/kg), starting 7 days before cell injection.
Tumor size was measured twice a week using digital calipers. Mice were separated into four groups (n = 7 mice/group): when tumors reached a volume of 150 mm3, they were treated or not with Sunitinib or Bevacizumab.
2.11. Bio‐Plex assay
Bio‐Plex human assay (Bio‐Rad Laboratories, Hercules, CA) for simultaneous quantitation of the angiogenic factor VEGF, was used according to the manufacturer's instructions. Briefly, supernatants of breast carcinoma cell lines seeded on plastic or Matrigel for 4 h were incubated on properly 96‐well plate with polystyrene‐beads coated with VEGF‐specific antibodies, and then exposed to detection antibodies prior incubation with Streptavidin‐PE. Capture lysates were analyzed on the Bio‐Plex™ 200 system (Bio‐Rad). Data are presented as concentration (pg/mL). The concentration of analyte bound to each bead is proportional to the median fluorescence intensity of reporter signal.
2.12. Statistical analysis
For experiments analysis data were compared using Student's t‐test and exact probability value for the relationship between two dichotomous variables was calculated by Fisher's exact test using Graph Pad Prism 5 software (GraphPad software Inc., San Diego, CA). All statistical tests were two‐sided at the conventional 5% significance level.
Disease Free Survival (DFS) cumulative incidence was defined as the time elapsed from the date of surgery to the date of the first event. DFS cumulative incidence curves were drawn by the life‐table method and the statistical significance tested by the log‐rank test. The cut‐off of Lacunae numbers was fixed at 75th percentile according to a preliminary analysis carried out on quartiles.
Analysis were conducted using SAS software (SAS Institute Inc., Cary, NC).
3. Results
3.1. TNBC cells acquire functional properties of endothelial cells
Analysis of tube formation assay on Matrigel revealed vascular channel formation in all six TN breast cancer cell lines, representative of different molecular categories (MDA‐MB‐468, HCC1937 and BT20 as basal‐like; MDA‐MB‐231, BT549 and MBA‐MB‐157 as mesenchymal‐like), even though only mesenchymal‐like TNBC cells were able to form completely closed loops, whereas no vascular structures were generated by luminal or HER2‐positive cells even at longer times (up to 24 h) (Figure 1A).
Figure 1.
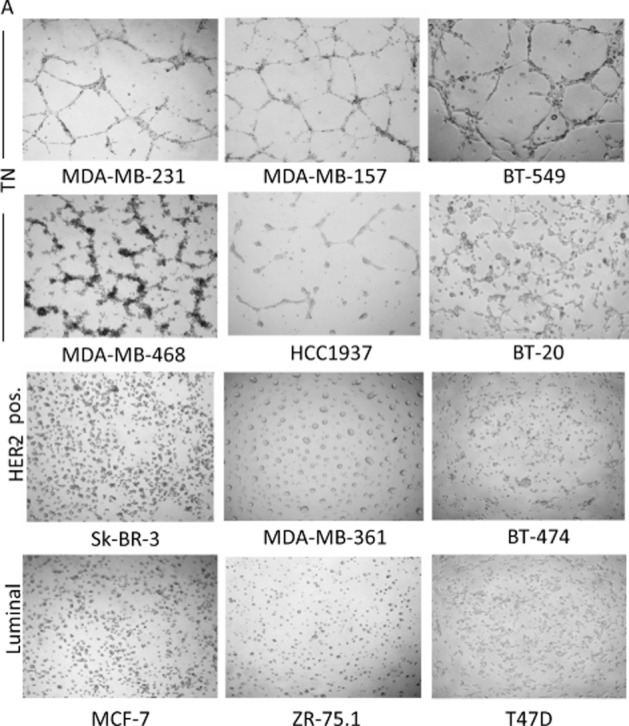
In vitro and in vivo VM. Tube formation capability was evaluated using a phase contrast light microscope (×100). Results are representative of five independent experiments (A). Histopathological analysis of engrafted tumors with MDA‐MB‐231 and MCF‐7 cells into mammary fat pad of SCID mice demonstrate that only TNBC neoplastic cells directly participate in tumor vascularization. i–iv. Vascular structures (lacunae) are generated in TNBC MDA‐MB‐231 xenografts. H&E staining (i, iii) shows the presence of nests (N) of atypical neoplastic cells in TNBC along with blood lacunae surrounded by tumor cells (arrows). Immunohistochemical assays with the endothelial marker CD31 (ii, iv) highlight that lacunae (asterisks) are composed of both endothelial and neoplastic cells (iv, inset). v–viii. MCF‐7 xenografts lack lacunae. Tumors present a nested and trabecular (T) pattern (v, vii) and have regular blood vessels exclusively composed of endothelial cells (vi,viii, arrows). Original magnification: i,ii,v,vi ×100; iii,iv,vii,viii ×200; inset ×400 (B).
Figure 1.
continued
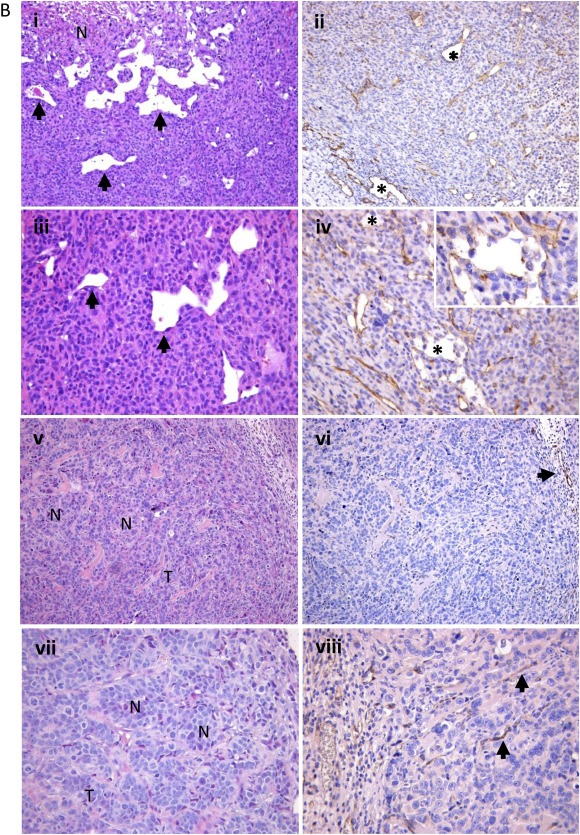
To determine whether the phenomenon of VM observed in vitro could actually reflect a property of TNBC cells also present in vivo, TN MDA‐MB‐231 or MDA‐MB‐468 and MCF‐7 cells were injected into mammary fat pad of SCID mice. The evaluation of formalin‐fixed paraffin‐embedded (FFPE) sections of engrafted tumors with both MDA‐MB‐231 or MDA‐MB‐468 cell lines revealed, particularly at the edge of infiltration, vascular lacunae delimited by neoplastic cells with an elongated morphology and nuclear features similar to those of neighboring, peri‐vascular, neoplastic cells (a representative example of MDA‐MB‐231 xenograft in Figure 1B, panels i and iii). Some of these lacunae were filled with red blood cells and leukocytes, providing their functional relationship with the systemic circulation. Consistently with the acquisition of an endothelium‐like phenotype, atypical tumor cells lining vascular lacunae showed the expression of the CD31 marker, which was highlighted by immunohistochemistry (Figure 1B, panels ii and iv). Vice versa, MCF‐7‐derived xenografts lack vascular lacunae (Figure 1B, panels v and vii), showing instead regular blood vessels exclusively composed of CD31+ endothelial cells (Figure 1B, panels vi and viii).
The in vivo vasculogenic potential of TNBC xenografts was also revealed by confocal analysis on frozen tumor sections of TN MDA‐MB‐231‐xenografted mice injected with Tomato lectin, which allowed detection of vascular structures connected with the systemic circulation. The presence of vascular structures surrounded by MDA‐MB‐231 cells expressing the epithelial marker EGFR confirmed the presence of functional vascular lacunae delimited by tumor cells (Figure 2A). Moreover, these data were further confirmed using xenograft models of MDA‐MB‐231‐GFP injected with Alexa Fluor 555 WGA: confocal microscopy analysis allowed a clear evaluation of the blood lacunae surrounded by GFP positive tumor cells (Figure 2B).
Figure 2.
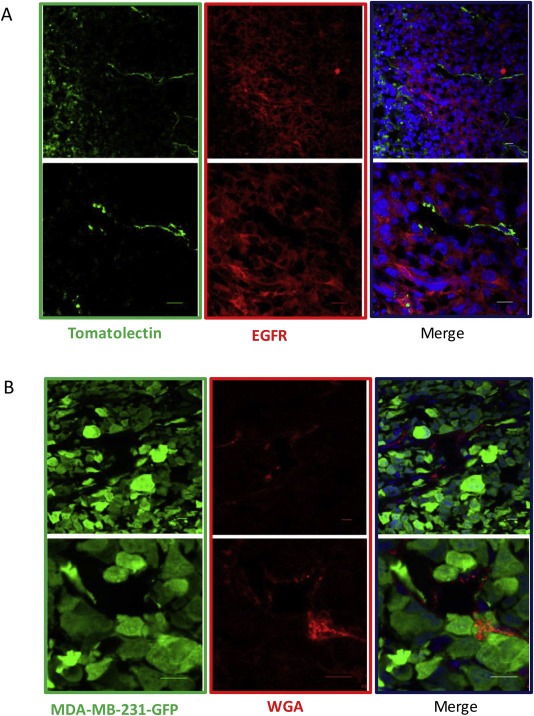
In vivo VM by confocal analysis. Images represent single frames of frozen tumor sections of tomato lectin‐injected MDA‐MB‐231 xenograft mice and show an example of channel‐like structures (green) delimited by tumor cells labeled with EGFR (red). Nuclei were stained with DRAQ‐5 (blue). Scale bars represent 10 μm (A). Images represent single frames of tumors deriving from MDA‐MB‐231‐GFP xenograft mice injected with Alexa Fluor 555 WGA and show blood lacunae (red) surrounded by GFP positive tumor cells (green). Nuclei were stained with DRAQ‐5 Scale bars represent 10 μm (B).
In addition, channel‐like structures formed by adjoining tumor cells tightly encompassing erythrocytes in xenotransplanted tumors deriving from MDA‐MB‐231 cells were also revealed by Transmission Electron Microscopy (TEM) analysis (Suppl. Figure 1A, left panel). Differently from TNBC‐derived xenografts, tumor masses from MCF‐7 engrafted mice lacked vascular lacunae, which were replaced by well‐structured vessels with recognizable lining endothelial cells (Suppl. Figure 1A, right panel).
We thus investigated the formation of tumor vascular lacunae in a series of 117 human breast cancer samples including 30 non‐TNBC (10 HER2 positive and 20 luminal) and 87 TNBC. The morphological evaluation of TNBC and non‐TNBC groups showed a different pattern of vasculature: whereas the non‐TNBC cases were mainly characterized by a canonical peripheral and intratumoral vascular network, composed by endothelial cell‐lined vessels, TNBC cases showed the formation of tumor vascular lacunae lined by neoplastic cells with a pseudo‐endothelial morphology and characteristic nuclear atypia (Figure 3A). Such lacunae were detected in 59 out of 87 (67.8%) TNBCs, 8 out of 20 (40%) luminal cases and in 3 out of 10 (30%) HER2 cases, and their number was assessed by counting the absolute number of vascular structures lined by neoplastic cells out of 5 high‐power fields (HPFs, ×400 magnification). Considering the cases showing VM, the median of the absolute number of vascular lacunae was significantly higher in TNBC cases group compared to luminal (p < 0.0001) or HER2 (p = 0.0055) group (Suppl. Figure S2). Also TEM evidenced channel‐like structures delimited by tumor cells and containing erythrocytes in 3 out of 5 TNBCs (an example is reported in Suppl. Figure 1B, left panel) but not in the two luminal breast tumors tested (Suppl. Figure S1B, right panel).
Figure 3.
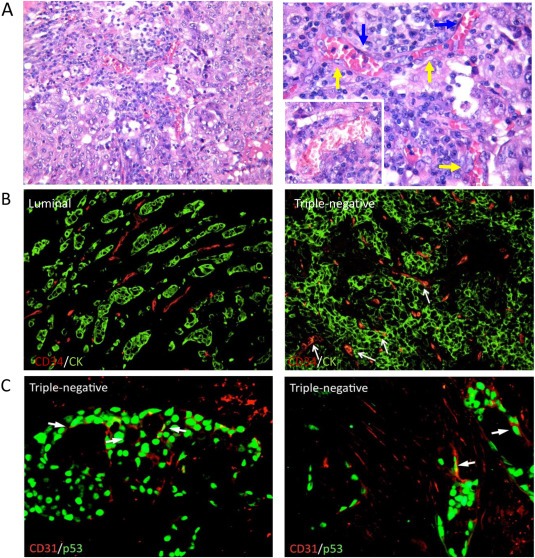
VM in human samples. Human TNBC samples showed the formation of tumor vascular lacunae lined by two different populations: one comprised large neoplastic cells with a pseudo‐endothelial elongated morphology and atypical nuclei (yellow arrows) and the other elongated regular endothelial cells (blue arrows). The lacunae are filled by erythrocytes and leukocytes. Original magnification ×200 (left panel), ×400 (right panel and inset) (A). Double‐immunofluorescent microscopy analysis of the CD34 endothelial marker and AE1/AE3 pan‐CK epithelial marker highlighted the expression of CD34 in CK+ neoplastic cells forming vascular structures in TNBCs. In non‐TNBC cases, CD34 and CK expression were not co‐localized. Original magnification ×200 (B). A double‐marker immunofluorescent analysis performed using CD31 and p53 specific antibodies revealed cells with elongated morphology lining the lacunae of TNBCs, further proving their neoplastic clone derivation. Original magnification ×400 (C).
Double‐immunofluorescent microscopy analysis of the CD34 endothelial marker and AE1/AE3 pan‐CK epithelial marker highlighted the expression of CD34 in CK+ neoplastic cells forming vascular structures in TNBCs. In non‐TNBC cases, CD34 and CK expression were not co‐localized (Figure 3B). This finding suggests the acquisition of an endothelial‐like phenotype, which impacts on both the morphology and immuno‐phenotype of TNBC cancer cells.
To further confirm the neoplastic epithelial origin of cells lining vascular lacunae in TNBC cases, a double‐marker immunofluorescent analysis was performed using CD31 and p53 specific antibodies. TNBCs are known to display mutated p53 isoforms which are detectable by immunostaining. The expression of p53 in CD31+ cells with elongated morphology lining the lacunae of TNBCs clearly proved their neoplastic clone derivation (Figure 3C).
Finally, to investigate the clinical impact of this biological phenomenon, we analyzed the DFS of TNBCs stratified according to the number of vascular lacunae, and found that the quartile with the highest number of lacunae is characterized by poorest prognosis (p = 0.055) (Figure 4). The Kaplan‐Meyer curve showing all quartiles is reported in Suppl. Figure S3.
Figure 4.
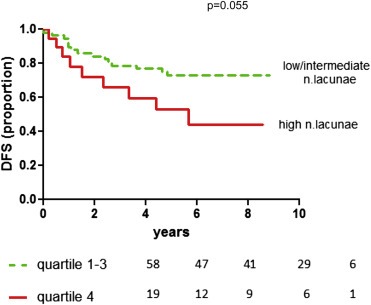
Kaplan‐Meyer curve reporting DFS of 77 TNBCs divided in two groups (one represented by the quartile characterized by the highest number of lacunae, the other group including all 3 other quartiles, which show a similar curve trend).
3.2. Pathways involved in endothelial‐like behavior of TNBC cells
To elucidate the mechanisms involved in VM, we investigated the possible involvement of molecules previously reported to be important in this phenomenon, as MMPs or EphA2 (Hendrix et al., 2003; Kirschmann et al., 2012). However, neither treatment with TIMP‐2 (MMP2 inhibitor) or Zometa (MMP9 inhibitor), at a concentration reported to be effective on VM inhibition in tumor cells (Hess et al., 2003) (Moschetta et al., 2010), or RNAi‐mediated EphA2 silencing (data not shown) produced any effect on the VM properties of our TN breast cancer cell lines.
Therefore, to evaluate whether the main pathways physiologically implicated in vessel formation by endothelial cells (i.e. VEGFR, PDFGR and/or FGFR) (Ferrara and Kerbel, 2005) could be responsible for this phenomenon, we used two different compounds, an antibody targeting VEGF (Bevacizumab) and a tyrosin‐kinase inhibitor targeting VEGFR, PDFGR, FGFR (Sunitinib). We first verified that MDA‐MB‐231 cells express these receptors by Real‐Time PCR (data not shown). Moreover, we assessed by Bioplex the production and secretion of VEGFA in the supernatant when cells are seeded in matrigel, and observed that its level is significantly higher in TN versus non‐TN cell lines (Suppl. Figure S4).
Treatment with Sunitinib almost completely abrogated the formation of vascular‐like channels in vitro in TNBC cell lines, whereas Bevacizumab did not show any inhibitory effect (Figure 5A, representative VM images reported in left panels and loop quantification in the right plot), suggesting that this phenomenon is likely to be dependent on FGFR and PDGFR‐induced pathways and does not rely on the autocrine VEGF/VEGFR loop.
Figure 5.
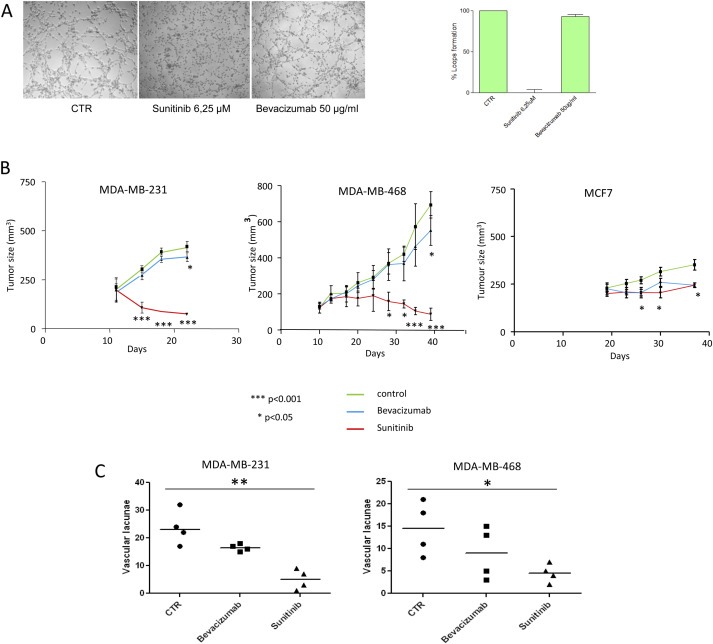
In vitro and in vivo effect of Sunitinib and Bevacizumab and quantification of “blood lacunae”. Cells were seeded in 96‐well tissue culture plates pre‐coated with Matrigel and treated with Bevacizumab or Sunitinib (A); the assay was quantified as percentage of loops formation compared to untreated cells (right panel). Each value represents the mean ± s.d. of data from three separate experiments. 5 × 106 TN MDA‐MB‐231, MDA‐MB‐468 or luminal MCF‐7 cells were injected into mammary fat pad of SCID mice, and when tumors reached a volume of 150 mm3, mice were treated or not with Sunitinib (per os, 40 mg/kg/day for 14 days) or Bevacizumab (i.p., 4 mg/kg twice at week for 4 weeks). Xenograft tumors derived from MDA‐MB‐231 and MDA‐MB‐468 cells showed highly significant responsiveness (***p < 0.001) to Sunitinib in comparison with MCF‐7 xenografts (*p < 0.05). Treatment with Bevacizumab caused comparable effects in terms of tumor volume reduction in MDA‐MB‐231, MDA‐MB‐468 and in MCF7‐derived xenograft mice. Error bars represent SD of 7 mice per group (B). Quantification of the absolute number of vascular structures lined by neoplastic cells out of 5 high‐power fields in sections from untreated, Sunitinib‐ or Bevacizumab‐treated MDA‐MB‐231 (left Scattered dot plot) or MDA‐MB‐468 (right Scattered dot plot) (*p = 0.0202, ***p = 0.0020) evaluated in four different mice (line at median value) (C).
We then decided to define whether PDGFR and FGFR mediated pathways might also influence proliferation of TNBC cells, in order to verify that VM inhibition was not related to cell growth impairment: in vitro growth of TNBC cell lines MDA‐MB‐231 and MDA‐MB‐468 and luminal MCF‐7 treated or not with Sunitinib or Bevacizumab was analyzed by SRB assay. Growth of all tested cell lines was not affected by Bevacizumab treatment, whereas sensitivity of TNBC cell lines to Sunitinib, evaluated calculating the IC50, was only slightly higher compared with luminal cells (approximately 5–6 μM in MDA‐MB‐468 and MDA‐MB‐231 cells, respectively, versus 10 μM in MCF‐7 cells at 72 h; Suppl. Figure S5).
To determine whether the inhibition of PDGFR and FGFR pathways had an impact on in vivo tumor growth, we evaluated the effect of Sunitinib and Bevacizumab in mouse models of MDA‐MB‐231, MDA‐MB‐468 and MCF‐7 cell lines.
MDA‐MB‐231 and MDA‐MB‐468 xenografts showed significantly higher responsiveness to Sunitinib (p < 0.001) compared with Bevacizumab (p < 0.05) (Figure 5B). Instead, in luminal MCF‐7‐derived xenograft mice, treatment with Sunitinib determined a tumor volume reduction comparable to the effect obtained with Bevacizumab (Figure 5B). Histomorphological evaluation reported in Figure 6 and quantified in Figure 5C showed that, even though also Bevacizumab was able to exert a partial inhibitory effect, only treatment with Sunitinib led to a statistically significant reduction in the number of vascular structures comprising tumor cells in TN xenograft mice (p = 0.0202 in MDA‐MB‐468; p = 0.0020 in MDA‐MB‐231).
Figure 6.
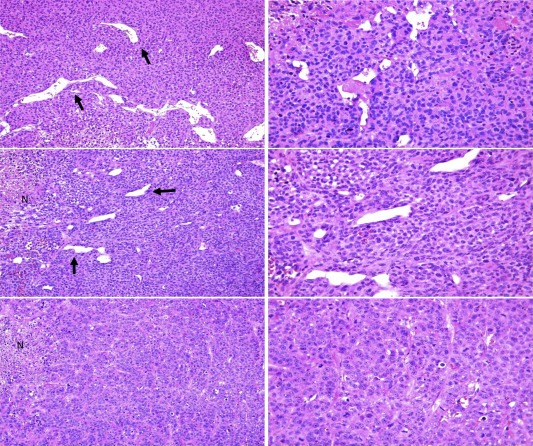
In vivo vascular lacunae. Differential density of vascular lacunae (black arrows) in untreated (upper panels), Bevacizumab (middle panels) and Sunitinib (lower panels) treated engrafted tumors. While control cases showed the higher presence of vascular lacunae, their number progressively decreases in Bevacizumab treated mice. Sunitinib treatment exerted the strongest effects by inducing the decrease of vascular lacunae. Focal areas of necrosis (N) could be detected in both Bevacizumab‐ or Sunitinib treated mice. Original magnification ×100 left panels, ×200 right panels.
However, since as above indicated Sunitinib is a multitarget agent, to better discriminate the contribution of the different molecules affected by this TKI, we used two additional compounds with a more specific effect: PD173074, a pan‐FGFR inhibitor, and Imatinib, TKI against PDGFR. As expected, treatment with both compounds strongly inhibited the capability to create vascular structures in vitro, recapitulating Sunitinib effect, whereas single agent treatment only partially affected the VM potential (Figure 7A, representative images in the left panel and loop quantification in the right plot), thus indicating that this property is likely to be dependent on both FGFR and PDGFR‐signaling pathways.
Figure 7.
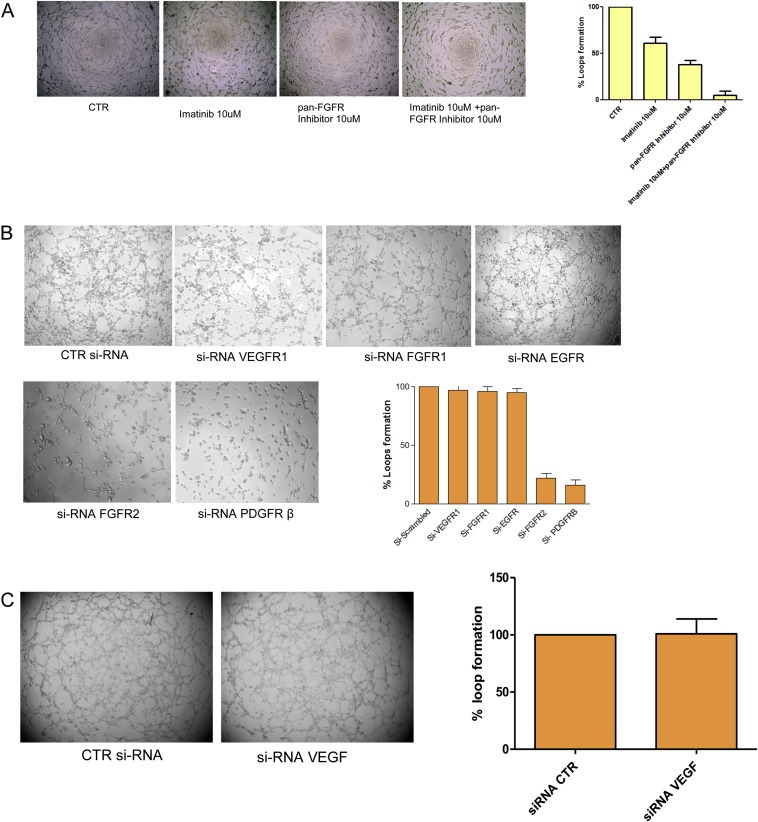
In vitro effect of pan‐FGFR inhibitor and Imatinib and siRNAs. Cells were seeded in 96‐well tissue culture plates pre‐coated with Matrigel and treated with pan‐FGFR inhibitor or Imatinib or both (10 uM each one) (A) for 4 h; for silencing experiments, cells previously transfected with specific siRNAs for 24–48 h were tested for VM for 4 h (B and C). The assay was quantified as percentage of loops formation compared to untreated cells. Each value represents the mean ± s.d. of data from three separate experiments.
To further investigate this hypothesis, and to deeper dissect the molecular pathways involved in this phenomenon, we silenced single components of the relevant networks by RNAi approach and evaluated the effect on VM. In detail, transient silencing of PDGFRβ or FGFR2 significantly inhibited the formation of channel structures of MDA‐MB‐231 cells, whereas no effect was observed using siRNA against FGFR1, EGFR or VEGFR1 (Figure 7B) and VEGF (7C), providing additional evidence of the marginal role of these pathways in mediating VM. The efficient silencing of the molecules of interest was evaluated by quantitative RT‐PCR (Suppl. Figure 6A), and evinced the possible existence of a crosstalk between FGFR2 and PDGFRβ, since even using single specific siRNA oligonucleotides directed against one receptor, we affected the expression of both.
Flow cytometric analysis of cell‐cycle progression performed after 24–48 h upon silencing showed approximately 20% decrease of S cell cycle phase in MDA‐MB‐231 cells treated with PDGFRβ (20%, p < 0.05) and FGFR2 (25%, p < 0.05) siRNAs. Similar results were obtained using MDA‐MB‐468 cells (20% with PDGFRβ, p < 0.01; 25% with FGFR2, p < 0.05) (Suppl. Figure 6B).
Since inhibitors targeting the single receptors (anti‐FGFR and Imatinib) produced a modest inhibitory effect on VM in comparison with specific siRNAs (Figure 7A), we repeated the treatment applying the same schedule used for siRNA transfection. Pre‐treatment for 24 h with single inhibitors (Imatinib or anti‐FGFR) significantly impaired VM potential of MDA‐MB‐231 cells (Suppl. Figure 7A, and quantified in right panel). As expected, impairment of FGFR and PDGFR pathways led to reduced expression of the other receptor (PDGFRβ and FGFR2, respectively), whereas no reduction was observed at short time (4 h) (Suppl. Figure 7B).
4. Discussion
Our study has highlighted the ability of TNBC cells to directly contribute to tumor vasculature by endothelial cell differentiation, a capability which recapitulates the plasticity of embryonic mesodermal progenitor cells performing the vasculogenic process in order to provide blood supply. Indeed, VM probably serves as a selective advantage to rapidly growing tumors by providing a perfusion pathway which contributes to the disease progression with the acquisition of a more aggressive and invasive phenotype. This hypothesis is supported by the evidence that, within the TNBC subgroup, a higher number of vascular lacunae is significantly associated with reduced DFS.
Even though the presence of vascular lacunae also in HER2 positive and luminal specimens might suggest that this phenomenon is not unique of TNBC, however it is certainly more common and peculiar, since it is present in a significantly higher percentage of cases and with a higher number of structures.
The correlation between VM and an undifferentiated phenotype reminiscent of embryologic events has been confirmed by the ability of cancer stem cells to perform this phenomenon (Ricci‐Vitiani et al., 2010), and very recently by the association with the expression of stemness marker CD133 in TNBC (Liu et al., 2012). At a functional point of view, our study demonstrates that a crucial role in mediating vasculogenic properties of TNBC is exerted by PDGFRβ and FGFR2 mediated pathways. Indeed, even though tested TNBC models do not present amplification or overexpression of these molecules, their inhibition strongly impairs the capability to form channel‐like structures, thus suggesting the involvement of these pathways in the observed phenomenon. It has been reported that MDA‐MB‐231 mainly express the beta isoform of PDGFR (Weigel et al., 2009) and the FGFR2 IIIc isoform, associated with a more mesenchymal and undifferentiated phenotype (Cha et al., 2009; Luqmani et al., 1996). Amplification of FGFR2 has been reported in a percentage of TNBCs and associated with poor prognosis (Dey et al., 2010), and PDGFRβ inhibition has been shown to impair the establishment of experimental bone (Farmer et al., 2005; Skehan et al., 1990) or lung (Jechlinger et al., 2006) metastases from breast cancer cells.
Even though we cannot completely exclude possible off targets effects of a multi‐target TKI as Sunitinib, the concentrations we used in our in vitro assays are consistent with previous reports, as a very recent work by Hollier et al. (2013), who used this compound to inhibit FOXC2‐induced PDGFRβ. Moreover, the impairment of VM upon specific RNAi‐mediated silencing of the receptors clearly demonstrates their involvement in this phenomenon.
Notably, the evidence that, even though we used very specific siRNA oligos, the silencing of FGFR2 partially affected also the expression of PDGFRβ, and vice versa, suggests the existence of a functional crosstalk, as already suggested in literature (Nissen et al., 2007). Moreover, the effect of 24 h pre‐treatment with single inhibitors (Imatinib and anti‐FGFR) on VM potential and on the expression of the other receptor (FGFR2 and PDGFRβ, respectively) further supports this hypothesis, and underlines that PDGFRβ and FGFR2 are probably both required in sustaining VM phenomenon.
Besides the in vitro studies, results of Sunitinib treatment in TN xenografts indicate that TN cancer cells, coherently with their endothelial features, strongly rely on PDGFRβ and FGFR2‐dependent pathways for their fitness and survival, as suggested by the impressive TN tumor growth inhibition paralleled by a conspicuous impairment of TN tumor vasculogenic potential.
Even though it has been reported that inhibition of VEGF reduces angiogenesis and growth of TN MDA‐MB‐231‐derived xenografts (Roland et al., 2009), in our hands the effect in terms of volume reduction mediated by Bevacizumab was definitely weaker than Sunitinib and comparable to the inhibition observed in luminal MCF‐7‐derived xenograft tumors. Surprisingly, and despite it does not affect VM in vitro, also Bevacizumab partially reduced the number of vascular lacunae, even though to a lesser extent and with no statistical significance: this capability might account for the slight but still significant reduction in xenograft tumor growth upon Ab treatment, and underlines how the in vivo setting can reveal mechanisms that we can underestimate by in vitro experiments. However, considering the strongest and statistically significant effect of Sunitinib in this phenomenon, we can certainly hypothesize that Bevacizumab is likely to act mainly on “standard angiogenesis”, characterized by the presence of endothelium‐surrounded blood vessels, whereas Sunitinib may target the pathways triggering the ability of tumor cells to behave as endothelial cells.
It cannot be excluded that these pathways are also involved in the proliferation or survival of TNBC cells, however the slight different sensitivity to Sunitinib showed by TNBC cells in comparison with our luminal model in vitro definitely cannot account for the impressive tumor growth inhibition observed in vivo only in TN‐derived xenograft mice. Moreover, the evidence that the conspicuous inhibition of the vasculogenic potential upon Sunitinib treatment of TN xenografts was not paralleled by a significant induction of cancer cell apoptosis (data not shown) further supports the hypothesis that these pathways are mainly responsible of driving the vasculogenic properties of TNBC, and that TN tumor growth inhibition is mainly due to the impairment of this phenomenon.
Moreover, it is also worth reporting that, when we performed VM assay, cells on matrigel were exposed to the drug for a very short period of time (4 h), and the minimum concentration we found to almost completely abrogate tube formation did not affect cell viability, thus excluding that the inhibitory effect in performing vascular structures might be due to cell suffering or proliferation arrest. Finally, evaluation of cell cycle progression of MDA‐MB‐231 and MDA‐MB‐468 cells at 24–48 h upon RNAi‐mediated silencing of PDFGRβ and FGFR2 showed a statistically significant but not impressive reduction in the percentage of S phase cells.
Despite the more efficient capability to perform VM in vitro showed by mesenchymal cell lines, our in vivo data show that both basal‐like (MDA‐MB‐468) and mesenchymal‐like (MDA‐MB‐231) TNBC cells contribute to tumor vascularization, and that Sunitinib strongly inhibits tumor growth of both TN models, thus indicating that inhibition of pathways involved in TNBC vasculature might represent an effective common therapy in this BC subtype. This aspect is extremely important, considering that the high molecular heterogeneity of TNBC, currently well recognized, has not been clearly elucidated yet.
However, the clinical application of Sunitinib is certainly limited by its relevant toxicity. Moreover, whereas Dey et al. (2010) reports that long term treatment with TKI258, targeting the same receptors inhibited by Sunitinib, induces both primary tumor growth and lung metastases reduction in a model of mammary carcinoma, two recent papers underline how short term treatment with anti‐angiogenic drugs as Sunitinib reduces the volume of the primary tumor but increases the number of metastasis in breast cancer (Ebos et al., 2009), glioma and pancreatic cancer (Paez‐Ribes et al., 2009). Our preliminary data also suggest an increase in the number of lung metastases upon Sunitinib treatment (data not shown).
The reason might rely on side effects on the tumor environment, probably mediating hypoxia and mobilitating tumor cells to intra‐vasate, and/or producing a severe vessel unbalance that facilitates extravasation and colonization at the metastatic site. This evidence, even though still controversial, limited to mouse models, and yet to be supported by a biological explanation, rises however a clearly important issue.
Considering the strong inhibitory effects on tube formation in vitro that we obtained silencing each single receptor, an alternative, or probably even more efficient, approach to a multitarget TKI as Sunitinib could be represented by an RNAi‐mediated therapy. However, it is of extreme importance to verify whether the silencing of these receptors is able to reduce the primary tumor growth without dangerously increasing the number of metastases (on going).
5. Conclusions
TNBC cells have the ability to form vascular‐like channels both in vitro, when seeded on Matrigel, and in vivo, generating blood lacunae lined by tumor cells. This peculiar endothelial behavior of TNBC, indicating an extremely undifferentiated phenotype, significant cell plasticity and the possibility to actively participate in providing blood supply, seems to contribute to the aggressiveness of this breast cancer subgroup, since a higher number of vascular structures is associated with poorer prognosis. Moreover, PDGFRβ‐ and FGFR2‐mediated pathways, identified as relevant in mediating this characteristic, potentially represent valid targets for a specific therapy of this breast cancer group.
Competing interests
The authors have no competing interests to declare.
Authors contributions
IP and MVI performed most part of the experiments and participated in both study design and manuscript writing; ET conceived, designed and coordinated the study, and supervised manuscript writing; ErD set tube formation assay in vitro; ElD contributed to siRNA experiments and evaluation; MS and MT carried out in vivo experiments; CaG, MatC and PC were responsible for IHC and IF experiments and analysis; CT and PC evaluated IHC and IF experiments and contributed in discussing and writing the manuscript; FB performed the Bioplex assay; TT helped with statistical analyses; CG helped with in vitro cultures; IB performed and evaluated TEM analysis; RA provided human samples; AB and ManC helped in discussing the study.
All authors read and approved the final manuscript.
Authors information
ET, head of Molecular Targeting Unit at the Fondazione IRCCS Istituto Nazionale Tumori of Milan, has a long experience and certified career on breast cancer research. She has an exhaustive knowledge of the molecular mechanisms driving breast cancer biology and resistance to therapy, with a particular focus on the understanding of the interactions between tumor and extracellular matrix.
MVI has an internationally recognized expertise on breast cancer, with a particular interest on microRNAs, and she is currently mainly focused on triple negative breast cancer.
CT is an internationally recognized professor of Pathological Anatomy.
PC has a long experience on immunofluorence, indeed she now leads the Confocal facility at the Istituto Nazionale Tumori of Milan.
ManC, Research Assistant, has recently won an AIRC Grant on triple negative breast cancer.
Supporting information
The following are the supplementary data related to this article:
Supplementary Figure 1 Representative images obtained using TEM analysis (×6000) of MDA‐MB‐231 (left panel) and MCF‐7 (right panel) xenograft tumors (A) and of human specimens, TNBC (left panel) and luminal (right panel) (B). The images show channel‐like structures delimited by tumor cells and containing erythrocytes (asterisks) in TNBC samples, whereas the luminal samples are characterized by small blood vessels containing an erythrocyte in the lumen surrounded by the endothelial lining.
Supplementary Figure 2 Scattered Dot plot of absolute number of vascular lacunae out of 5 high‐power fields in TN, luminal and HER2 human BC samples. Line at median value (**p = 0.0055, ***p < 0.0001).
Supplementary Figure 3 Kaplan‐Meyer curve reporting DFS of 77 TNBCs divided in quartiles according to the number of vascular lacunae.
Supplementary Figure 4 Supernatants recovered from MDA‐MB‐231, MDA‐MB‐157, MDA‐MB‐468, HCC1937, SKBr3, MDA‐MB‐361, MCF‐7 and ZR75.1 breast cancer cell lines seeded on plastic dishes pre‐coated with Matrigel for 4 h were analyzed for VEGF concentrations by Bio‐Plex assay and an unpaired t‐test was performed comparing VEGF concentration in supernatants from TNBC cells or from non‐TNBC.
Supplementary Figure 5 SRB assay was performed to evaluate IC50 of Sunitinib (upper panel) or Bevacizumab (lower panel) at 72 h in MDA‐MB‐231, MDA‐MB‐468 and MCF‐7 cells.
Supplementary Figure 6 The expression levels of the molecules of interest following siRNA transfection was evaluated by Real‐Time PCR (A) (each value is the average of three different replicates, error bars represent SD); and the effect on cell cycle at 48 h is reported in (B) (each value is the average of two technical replicates, error bars indicate SD, and the reported histogram is representative of three different experiments). PDGFRβ (F: 5′‐AGCGCTGGCGAAATCG‐3′; R: 5′‐TGACACTGGTTCGCGTGAA‐3′); FGFR2 (F: 5′‐AGATTGAGGTTCTCTATATTCGGAAT‐3′; R: 5′‐TTCTCTTCCAGGCGCTGG‐3′); FGFR1 (F: 5′‐AGGTGCTTCACTTAAGAAATGTCTCC‐3′; R: 5′‐GCCTCTCTTCCAGGGCTTC‐3′) VEGFR1 (F: 5′‐TCCCTTATGATGCCAGCAAGT‐3′; R: 5′‐CCAAAAGCCCCTCTTCCAA‐3′) and GAPDH (F: 5′‐ATTCCACCCATGGCAAATTC‐3′; R: 5′‐AGCATCGCCCCACTTGATT‐3′) were used with Sybr‐Green; for EGFR and VEGF detection we used Taqman probe.
Supplementary Figure 7 In vitro effect of pan‐FGFR inhibitor and/or Imatinib at 24 h. Cells pre‐treated for 24h with pan‐FGFR inhibitor or Imatinib or both (10 uM each one) were seeded on matrigel as previously described and tested for VM (A). The assay was quantified as percentage of loops formation compared to untreated cells. Each value represents the mean ± s.d. of data from three separate experiments. 24 h pre‐treatment with single inhibitors (Imatinib or anti‐FGFR) affects the expression of the other receptor (FGFR and PDGFRβ, respectively), whereas no reduction was observed at short time (4 h) (B).
Acknowledgments
Thank to Dr. Abbate for elaborating an algorithm with Image ProPlus able to recognize and complete loops in the Vasculogenic Mimicry assay, Dr. Colombo MP for providing MDA‐MB‐231‐GFP cells, Dr. Alessandro Gulino for excellent technical assistance and Mrs. Mameli L. for helping in figure editing.
This work is partially supported by AIRC and by Italian Ministry of Health.
Dr. Iorio is supported by an START UP AIRC grant and by Italian Ministry of Health.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.03.015.
Plantamura Ilaria, Casalini Patrizia, Dugnani Erica, Sasso Marianna, D'Ippolito Elvira, Tortoreto Monica, Cacciatore Matilde, Guarnotta Carla, Ghirelli Cristina, Barajon Isabella, Bianchi Francesca, Triulzi Tiziana, Agresti Roberto, Balsari Andrea, Campiglio Manuela, Tripodo Claudio, Iorio Marilena V. and Tagliabue Elda, (2014), PDGFRβ and FGFR2 mediate endothelial cell differentiation capability of triple negative breast carcinoma cells, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.03.015.
Contributor Information
Ilaria Plantamura, Email: ilaria.plantamura@istitutotumori.mi.it.
Patrizia Casalini, Email: patrizia.casalini@istitutotumori.mi.it.
Erica Dugnani, Email: erica.dugnani@istitutotumori.mi.it.
Marianna Sasso, Email: marianna.sasso@istitutotumori.mi.it.
Elvira D'Ippolito, Email: elvira.dippolito@istitutotumori.mi.it.
Monica Tortoreto, Email: monica.tortoreto@istitutotumori.mi.it.
Matilde Cacciatore, Email: matilde.cacciatore@unipa.it.
Carla Guarnotta, Email: carla.guarnotta@unipa.it.
Cristina Ghirelli, Email: cristina.ghirelli@istitutotumori.mi.it.
Isabella Barajon, Email: isabella.barajon@unimi.it.
Francesca Bianchi, Email: francesca.bianchi@istitutotumori.mi.it.
Tiziana Triulzi, Email: tiziana.triulzi@istitutotumori.mi.it.
Roberto Agresti, Email: roberto.agresti@istitutotumori.mi.it.
Andrea Balsari, Email: andrea.balsari@unimi.it.
Manuela Campiglio, Email: manuela.campiglio@istitutotumori.mi.it.
Claudio Tripodo, Email: claudio.tripodo@unipa.it.
Marilena V. Iorio, Email: marilena.iorio@istitutotumori.mi.it
Elda Tagliabue, Email: elda.tagliabue@istitutotumori.mi.it.
References
- Cha, J.Y. , Maddileti, S. , Mitin, N. , Harden, T.K. , Der, C.J. , 2009. Aberrant receptor internalization and enhanced FRS2-dependent signaling contribute to the transforming activity of the fibroblast growth factor receptor 2 IIIb C3 isoform. J. Biol. Chem.. 284, 6227–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang, M.C. , Voduc, D. , Bajdik, C. , Leung, S. , McKinney, S. , Chia, S.K. , Perou, C.M. , Nielsen, T.O. , 2008. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res.. 14, 1368–1376. [DOI] [PubMed] [Google Scholar]
- Dawood, S. , Broglio, K. , Kau, S.W. , Green, M.C. , Giordano, S.H. , Meric-Bernstam, F. , Buchholz, T.A. , Albarracin, C. , Yang, W.T. , Hennessy, B.T. , Hortobagyi, G.N. , Gonzalez-Angulo, A.M. , 2009. Triple receptor-negative breast cancer: the effect of race on response to primary systemic treatment and survival outcomes. J. Clin. Oncol.. 27, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey, J.H. , Bianchi, F. , Voshol, J. , Bonenfant, D. , Oakeley, E.J. , Hynes, N.E. , 2010. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res.. 70, 4151–4162. [DOI] [PubMed] [Google Scholar]
- Di Leva, G. , Gasparini, G. , Piovan, C. , Ngankeu, A. , Garofalo, M. , Taccioli, C. , Iorio, M.V. , Li, M. , Volinia, S. , Alder, H. , Nakamura, T. , Nuovo, G. , Liu, Y. , Nephew, K.P. , Croce, C.M. , 2010. MicroRNA cluster 221-222 and estrogen receptor alpha interactions in breast cancer. J. Natl. Cancer I. 102, 706–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos, J.M. , Lee, C.R. , Cruz-Munoz, W. , Bjarnason, G.A. , Christensen, J.G. , Kerbel, R.S. , 2009. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell.. 15, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, I.O. , Elston, C.W. , 2006. Histologic grade. In O'Malley F.P., Pinder S.E.(Eds.), Breast Pathology. Elsevier; Philadelphia, PA: 225–233. [Google Scholar]
- Farmer, P. , Bonnefoi, H. , Becette, V. , Tubiana-Hulin, M. , Fumoleau, P. , Larsimont, D. , MacGrogan, G. , Bergh, J. , Cameron, D. , Goldstein, D. , Duss, S. , Nicoulaz, A.L. , Brisken, C. , Fiche, M. , Delorenzi, M. , Iggo, R. , 2005. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene. 24, 4660–4671. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. , Kerbel, R.S. , 2005. Angiogenesis as a therapeutic target. Nature. 438, 967–974. [DOI] [PubMed] [Google Scholar]
- Hendrix, M.J. , Seftor, E.A. , Hess, A.R. , Seftor, R.E. , 2003. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat. Rev. Cancer. 3, 411–421. [DOI] [PubMed] [Google Scholar]
- Hess, A.R. , Seftor, E.A. , Seftor, R.E. , Hendrix, M.J. , 2003. Phosphoinositide 3-kinase regulates membrane Type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry. Cancer Res.. 63, 4757–4762. [PubMed] [Google Scholar]
- Hollier, B.G. , Tinnirello, A.A. , Werden, S.J. , Evans, K.W. , Taube, J.H. , Sarkar, T.R. , Sphyris, N. , Shariati, M. , Kumar, S.V. , Battula, V.L. , Herschkowitz, J.I. , Guerra, R. , Chang, J.T. , Miura, N. , Rosen, J.M. , Mani, S.A. , 2013. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast Cancer. Cancer Res.. 73, 1981–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvin, W.J. , Carey, L.A. , 2008. What is triple-negative breast cancer?. Eur. J. Cancer. 44, 2799–2805. [DOI] [PubMed] [Google Scholar]
- Jechlinger, M. , Sommer, A. , Moriggl, R. , Seither, P. , Kraut, N. , Capodiecci, P. , Donovan, M. , Cordon-Cardo, C. , Beug, H. , Grunert, S. , 2006. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 116, 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschmann, D.A. , Seftor, E.A. , Hardy, K.M. , Seftor, R.E. , Hendrix, M.J. , 2012. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin. Cancer Res.. 18, 2726–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal, M. , Ell, B.J. , Buffa, F.M. , Ibrahim, T. , Blanco, M.A. , Celia-Terrassa, T. , Mercatali, L. , Khan, Z. , Goodarzi, H. , Hua, Y. , Wei, Y. , Hu, G. , Garcia, B.A. , Ragoussis, J. , Amadori, D. , Harris, A.L. , Kang, Y. , 2011. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med.. 17, 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, B.D. , Bauer, J.A. , Chen, X. , Sanders, M.E. , Chakravarthy, A.B. , Shyr, Y. , Pietenpol, J.A. , 2011. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T.J. , Sun, B.C. , Zhao, X.L. , Zhao, X.M. , Sun, T. , Gu, Q. , Yao, Z. , Dong, X.Y. , Zhao, N. , Liu, N. , 2012. CD133(+) cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 10, [DOI] [PubMed] [Google Scholar]
- Luqmani, Y.A. , Bansal, G.S. , Mortimer, C. , Buluwela, L. , Coombes, R.C. , 1996. Expression of FGFR2 BEK and K-SAM mRNA variants in normal and malignant human breast. Eur. J. Cancer. 32A, 518–524. [DOI] [PubMed] [Google Scholar]
- Ménard, S. , 2011. Heterogeneity of triple-negative breast carcinomas. Oncologie. 14, 28–30. [Google Scholar]
- Moschetta, M. , Di, P.G. , Ria, R. , Gnoni, A. , Mangialardi, G. , Guarini, A. , Ditonno, P. , Musto, P. , D'Auria, F. , Ricciardi, M.R. , Dammacco, F. , Ribatti, D. , Vacca, A. , 2010. Bortezomib and zoledronic acid on angiogenic and vasculogenic activities of bone marrow macrophages in patients with multiple myeloma. Eur. J. Cancer. 46, 420–429. [DOI] [PubMed] [Google Scholar]
- Nissen, L.J. , Cao, R. , Hedlund, E.M. , Wang, Z. , Zhao, X. , Wetterskog, D. , Funa, K. , Brakenhielm, E. , Cao, Y. , 2007. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J. Clin. Invest. 117, 2766–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes, M. , Allen, E. , Hudock, J. , Takeda, T. , Okuyama, H. , Vinals, F. , Inoue, M. , Bergers, G. , Hanahan, D. , Casanovas, O. , 2009. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell.. 15, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou, C.M. , 2011. Molecular stratification of triple-negative breast cancers. The Oncologist. 16, (Suppl. 1) 61–70. [DOI] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , Fluge, O. , Pergamenschikov, A. , Williams, C. , Zhu, S.X. , Lonning, P.E. , Borresen-Dale, A.L. , Brown, P.O. , Botstein, D. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Prat, A. , Perou, C.M. , 2011. Deconstructing the molecular portraits of breast cancer. Mol. Oncol.. 5, 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani, L. , Pallini, R. , Biffoni, M. , Todaro, M. , Invernici, G. , Cenci, T. , Maira, G. , Parati, E.A. , Stassi, G. , Larocca, L.M. , De, M.R. , 2010. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 468, 824–828. [DOI] [PubMed] [Google Scholar]
- Roland, C.L. , Dineen, S.P. , Lynn, K.D. , Sullivan, L.A. , Dellinger, M.T. , Sadegh, L. , Sullivan, J.P. , Shames, D.S. , Brekken, R.A. , 2009. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol. Cancer Ther.. 8, 1761–1771. [DOI] [PubMed] [Google Scholar]
- Skehan, P. , Storeng, R. , Scudiero, D. , Monks, A. , McMahon, J. , Vistica, D. , Warren, J.T. , Bokesch, H. , Kenney, S. , Boyd, M.R. , 1990. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer I. 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Weigel, M.T. , Meinhold-Heerlein, I. , Bauerschlag, D.O. , Schem, C. , Bauer, M. , Jonat, W. , Maass, N. , Mundhenke, C. , 2009. Combination of imatinib and vinorelbine enhances cell growth inhibition in breast cancer cells via PDGFR beta signalling. Cancer Lett.. 273, 70–79. [DOI] [PubMed] [Google Scholar]
- World Medical Association Declaration of Helsinki, 2013. ethical principles for medical research involving human subjects. JAMA. 310, 2191–2194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Figure 1 Representative images obtained using TEM analysis (×6000) of MDA‐MB‐231 (left panel) and MCF‐7 (right panel) xenograft tumors (A) and of human specimens, TNBC (left panel) and luminal (right panel) (B). The images show channel‐like structures delimited by tumor cells and containing erythrocytes (asterisks) in TNBC samples, whereas the luminal samples are characterized by small blood vessels containing an erythrocyte in the lumen surrounded by the endothelial lining.
Supplementary Figure 2 Scattered Dot plot of absolute number of vascular lacunae out of 5 high‐power fields in TN, luminal and HER2 human BC samples. Line at median value (**p = 0.0055, ***p < 0.0001).
Supplementary Figure 3 Kaplan‐Meyer curve reporting DFS of 77 TNBCs divided in quartiles according to the number of vascular lacunae.
Supplementary Figure 4 Supernatants recovered from MDA‐MB‐231, MDA‐MB‐157, MDA‐MB‐468, HCC1937, SKBr3, MDA‐MB‐361, MCF‐7 and ZR75.1 breast cancer cell lines seeded on plastic dishes pre‐coated with Matrigel for 4 h were analyzed for VEGF concentrations by Bio‐Plex assay and an unpaired t‐test was performed comparing VEGF concentration in supernatants from TNBC cells or from non‐TNBC.
Supplementary Figure 5 SRB assay was performed to evaluate IC50 of Sunitinib (upper panel) or Bevacizumab (lower panel) at 72 h in MDA‐MB‐231, MDA‐MB‐468 and MCF‐7 cells.
Supplementary Figure 6 The expression levels of the molecules of interest following siRNA transfection was evaluated by Real‐Time PCR (A) (each value is the average of three different replicates, error bars represent SD); and the effect on cell cycle at 48 h is reported in (B) (each value is the average of two technical replicates, error bars indicate SD, and the reported histogram is representative of three different experiments). PDGFRβ (F: 5′‐AGCGCTGGCGAAATCG‐3′; R: 5′‐TGACACTGGTTCGCGTGAA‐3′); FGFR2 (F: 5′‐AGATTGAGGTTCTCTATATTCGGAAT‐3′; R: 5′‐TTCTCTTCCAGGCGCTGG‐3′); FGFR1 (F: 5′‐AGGTGCTTCACTTAAGAAATGTCTCC‐3′; R: 5′‐GCCTCTCTTCCAGGGCTTC‐3′) VEGFR1 (F: 5′‐TCCCTTATGATGCCAGCAAGT‐3′; R: 5′‐CCAAAAGCCCCTCTTCCAA‐3′) and GAPDH (F: 5′‐ATTCCACCCATGGCAAATTC‐3′; R: 5′‐AGCATCGCCCCACTTGATT‐3′) were used with Sybr‐Green; for EGFR and VEGF detection we used Taqman probe.
Supplementary Figure 7 In vitro effect of pan‐FGFR inhibitor and/or Imatinib at 24 h. Cells pre‐treated for 24h with pan‐FGFR inhibitor or Imatinib or both (10 uM each one) were seeded on matrigel as previously described and tested for VM (A). The assay was quantified as percentage of loops formation compared to untreated cells. Each value represents the mean ± s.d. of data from three separate experiments. 24 h pre‐treatment with single inhibitors (Imatinib or anti‐FGFR) affects the expression of the other receptor (FGFR and PDGFRβ, respectively), whereas no reduction was observed at short time (4 h) (B).