

Abstract
Purpose:
Breast cancer is among the leading causes of cancer death; discovery of novel prognostic markers is needed to improve outcomes. Combining systems biology and epidemiology, we investigated microRNA-associated genes and breast cancer survival in a well-characterized population-based study.
Experimental Design:
A recently developed algorithm, ActMiR, was used to identify key microRNAs “activities” corresponding to target gene degradation which were predictive of breast cancer mortality in published databases. We profiled microRNA-associated genes in tumors from our well-characterized population-based cohort of 606 women with first primary breast cancer. Cox proportional hazards models were used to estimate hazard ratios (HR) and 95% confidence intervals (CI), after 15+ years of follow-up with 119 breast cancer-specific deaths.
Results:
miR-500a activity was identified as a key microRNA for estrogen receptor positive breast cancer mortality using public databases. From a panel of 161 miR-500a associated genes profiled, 73 were significantly associated with breast cancer-specific mortality (FDR<0.05) in our population, among which two clusters were observed to have opposing directions of association. For example, high level of SUSD3 was associated with reduced breast cancer-specific mortality [HR 0.3, 95% CI: 0.2-0.4], while the opposite was observed for TPX2 [HR 2.7, 95% CI: 1.8-3.9]. Most importantly, we identified set of genes for which associations with breast cancer-specific mortality were independent of known prognostic factors including hormone receptor status and PAM-50 derived Risk of Recurrence scores. These results are validated in independent datasets.
Conclusions:
We identified novel markers that may improve prognostic efficiency while shedding light on molecular mechanisms of breast cancer progression.
Introduction
Recent advances in molecular phenotyping of breast cancer have significantly advanced our capabilities to treat breast cancer based on molecular features of the tumor and ultimately to improve survival (1). Different gene expression panels, such as Oncotype DX™, MammaPrint™ and Prosigna™, have been developed and approved for clinical use for predicting risk of recurrence for early stage breast cancer (2). Using different platforms and detection techniques, these panels allow relatively robust prediction of the risk of breast cancer relapse in the short term (0-5 years), but lower prediction power for long-term follow-up (3). Development of a risk score plays an important role in choosing treatment modality: high-risk patients typically undergo more aggressive chemotherapy, while low-risk patients fare well with hormonal treatment alone (4). While the treatment regimen for high- and low-risk groups is chosen with a high degree of certainty, the choice of treatment for the patients with intermediate risk is more difficult; heterogeneity of outcome within this group means a certain number of patients will be misclassified (5). Thus there is urgent need to identify additional molecular markers that can refine the risk prediction for long-term survival. Given that currently approved diagnostic gene panels involve between 21 and 70 genes, it would be beneficial to develop a simpler prognostic panel with a smaller number of genes to better characterize breast cancer mortality risk.
MicroRNAs are known as “universal regulators” of gene expression and are involved in post-transcriptional regulation of multiple fundamental cellular processes in cancer pathophysiology including cell proliferation, differentiation, apoptosis and the stemness of the cancer cells. These processes in turn, can be linked to tumor initiation, progression, and metastasis (6). Because their mechanism of action involves limited complementarity, each miRNA can potentially repress hundreds of mRNA targets. It is now clear that miRNAs are involved in virtually all signaling pathways including those crucial for cell transformation and cancer progression (6). It is thus not surprising that recent studies have attempted to include miRNAs as potential biomarkers of progression (7). However, miRNA expression level is not equivalent to its functional activity (8). Instead, the relative abundance of miRNAs to its target genes determines the functional activity levels of miRNAs (9). We have recently reported a novel computational approach, ActMiR, for identifying active miRNAs and miRNA-mediated regulatory mechanism and have shown stronger associations between the “activity” of miRNAs with clinical outcomes (including breast cancer survival) compared to the level of expression (10). For example, we identified that high activities of let-7d and miR-18a were associated with better survival outcomes for estrogen receptor negative human epidermal growth factor receptor 2 negative (ER−/HER2−) breast cancer. As we report in the current study, the same ActMiR algorithm also suggests that high miR-500a activity would be associated with poor survival among ER+ breast cancer patients. Because miR-500a activity cannot be directly measured but only inferred through expression of its associated genes, we designed the translational study reported here to investigate the association of miR-500a mediated gene expression with long-term breast cancer survival.
Materials and Methods
Study population
We utilized resources from the follow-up component of the Long Island Breast Cancer Study Project (LIBCSP), which includes a population-based cohort of women newly diagnosed with first primary breast cancer who participated in the baseline interview within two to three months of diagnosis (11) and were subsequently re-interviewed about five years later and followed for vital status (12). Participants were 1508 women newly diagnosed with first primary breast cancer between August 1, 1996 and July 31, 1997, who resided in Nassau and Suffolk counties of Long Island, NY, predominantly (90%) Caucasian, and aged 25-98 (median age 58). Potentially eligible women were identified through daily or weekly contact with pathology/cytology departments of 33 collaborating institutions, and confirmed by medical record. The National Death Index (NDI) was used to ascertain all-cause and breast cancer-specific mortality among study participants, who were followed from diagnosis until December 31, 2014, for an average of 162 months (range 2.7 – 224 months, median 213 months). We obtained sufficient RNA for profiling from paraffin-embedded tumor tissue from 616 of the 1508 patients (for the other LIBCSP patients, tumor slides were not made available by the diagnosing hospitals (n=763) or the extracted RNA was of insufficient/low quality (n=139); of these, 606 further passed QC. Demographic description of both the overall patient cohort and the subset with sufficient tissue RNA is presented in Table S1. Among the 606 women with successfully profiled tumor tissue, 275 deaths occurred by the end of the follow-up period, of which 119 (43%) were related to breast cancer (ICD codes 174.9 and C-50.9 listed as a primary or secondary code on the death certificate). The study protocol was approved by the Institutional Review Boards of the collaborating institutions.
Activity of microRNAs
We recently developed a computational method, ActMiR, which infers miRNA activity based on expression levels of miRNAs and their potential target genes (10). In brief, three pieces of information are used in this method: (i) a list of predicted targets of a given miRNA; (ii) expression levels of this miRNA samples of interest; (iii) mRNA expression levels of predicted targets in the same samples. This approach infers “functional activity” of a given miRNA using a regression-based model based on the expression levels of all miR predicted target genes. For the predicted target list of miRNAs, we used a collection of predicted target genes for 1537 unique mature miRNAs from TARGETSCAN (www.targetscan.org) that considers all conserved miRNA binding sites inherited from 23-way alignments of UTR sequences (13). In order to obtain robust results, we filtered out miRNAs whose number of target genes was smaller than 10. High miRNA activity corresponds to the high degradation effect on their target genes. To identify microRNAs of which activity was associated with breast cancer survival, we used “The Cancer Genome Atlas” database (tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp) and data described in Dvinge et al. (14).
Selection of miR-500a mediated genes
We previously published a novel computational method, ActMiR, for inferring microRNA activity (10). High miR-500a “activity” was predicted to be associated with increased ER+ breast cancer mortality (see Supplementary Methods). Because the “activity” of miR-500 can only be inferred by expression of its mediated genes, we selected miR-500a mediated genes to be profiled in the LIBCSP tumor samples. They included genes that were significantly correlated with imputed miR-500a activity and also those differentially expressed after modulation of miR-500a in breast cancer cells. A total of 161 miR-500a mediated genes were selected. We also included all genes of the PAM50 panel by NanoString (15) in order to take into account of known risk-of-recurrence factors; 22 genes of the PAM50 set overlapped with the miR-500a-mediated gene set. Lastly, 6 genes were used as housekeeping controls and for normalization: CLTC, GAPDH, GUSB, HPRT1, PGK1 and TUBB. These bring the total number of genes profiled to 195; the complete list is shown in Table S2.
Tissue processing and RNA extraction
Formalin-fixed paraffin-embedded (FFPE) tumor sections from the population-based cohort were histopathologically reviewed by a trained pathologist and the cancer tissue was separated using manual microdissection (16). Total RNA was extracted from tumor tissues using the Qiagen miRNeasy FFPE kit (Qiagen, MD). RNA concentration and quality were determined with a NanoDrop spectrophotometer (Thermo Scientific, MA). In total, 606 samples with sufficient RNA concentration were used in our analyses.
Gene expression profiling
Gene expression analysis was performed with the NanoString nCounter platform (Seattle, WA), using a custom-designed codeset and following the manufacturer’s instructions. Briefly, 100 ng RNA was incubated with reporter and capture probes overnight at 65°C. Following hybridization, unbound probes were removed, and the purified complexes were aligned and immobilized on imaging cartridges using an nCounter Prep station. Code count detection was carried out by scanning cartridges in an nCounter Digital Analyzer to determine gene expression levels. As a quality control filter, we excluded all samples having less than 50000 total counts for positive probes, or having less than 1.5*blank counts for housekeeping and endogenous genes. Raw counts were imported from RCC files with the NanoStringQCPro R package (version 1.6.0), background was subtracted as mean of negative controls counts, geometrical mean of housekeeping genes counts was used for normalization, and values were log-transformed for further analysis.
Coexpression of profiled genes was analyzed in R by building a covariance matrix; principal component analysis was performed with “pca” function of “pcaMethods” package (17).
Molecular characteristics of tumors and risk-of-recurrence score
We were able to obtain hormone receptor (HR) status determined by immunohistochemistry (IHC) from medical records for 475 out of 606 profiled samples, among which 75% and 62% were positive for estrogen receptor (ER+) and progesterone receptor (PR+), respectively. In addition, mRNA levels corresponding ESR1 and PGR were measured by NanoString in all tumor samples. For analyses in this study, we redefined ER/PR status based on ESR1/PGR mRNA expression levels (see Supplementary Methods). The expression-based method resulted in prevalence of 78% for ER+ (kappa=0.77) and 67% for PR+ (kappa=0.66) breast tumors (Fig. S1, Fig. S2).
Recently, more advanced prognostic tools based on larger gene panels, such as PAM50-derived “Prosigna™” , have been approved for clinical use and shown to improve breast cancer survival (18). In this study, we profiled expression of PAM50 (15) genes and determined molecular subtype and risk-of-recurrence (ROR) score using R code of Joel S. Parker from https://genome.unc.edu/pubsup/breastGEO/PAM50.zip archive (15). Risk-of-recurrence score was calculated in the “Subtype + Proliferation” variant.
Survival analysis
Cox proportional hazards models (19) were used for estimating the multivariate-adjusted hazard ratios (HRs) and 95% confidence intervals (CIs) for breast cancer-specific mortality associated with gene expression among the 606 women in our population-based cohort. Survival analysis was performed using “survival” package in R (20). For each gene, range of cutoff thresholds (from second to eighth decile) was tested and the threshold with the lowest p-value was used (21). For each gene, a Cox model was built using coxph function with age and tumor stage (invasive or in situ) included as cofactors. Multiple comparison adjustment was applied, where indicated, with the false discovery rate (FDR) threshold at 0.05.
All models were initially adjusted for age at diagnosis (continuous) and tumor stage. We further evaluated potential confounding using the methods described by Rothman and Greenland (22) starting with a full multivariate model and using backward elimination. Potential confounder considered included: menopausal status (pre-/post-menopausal), family history of breast cancer in a first-degree relative, body mass index (BMI) at diagnosis, tumor size and education. If eliminating a covariate from the full Cox regression model changed the effect estimate by 10% or more, the covariate was considered a confounder and kept in the model. Otherwise that covariate was dropped from the multivariate model. None of the covariates tested met such criterion, thus, only results adjusted for age and tumor stage were presented.
We also performed stratified analyses by ER/PR status as well as ROR scores discussed above. Kaplan-Meier survival (23) plots were built for visualization of unadjusted associations between gene expression and breast cancer-specific mortality.
To estimate receiving operator characteristics (ROC), we first used R package pROC (24). This package allows to perform a standard ROC analysis and calculates the Area Under the Curve (AUC) for binary outcome, i.e. censored (death by breast cancer) vs. uncensored (survived at the end of followup). However, this is not the optimal estimator for such time-dependent variable as patients survival, therefore we further re-analyzed our data using the survivalROC package (25). This package takes into account the survival duration and is considered more appropriate for this type of data (25). We used Nearest Neighbor Estimation (NNE) variant of the survivalROC function, with time point of 365 days and span parameter of 0.07.
Results
Activity of miR-500a and breast cancer mortality
The large public cancer dataset, the Cancer Genome Atlas (TCGA), contains multiple types of data (e.g. DNA copy number arrays, DNA methylation, exome sequencing, messenger RNA array, microRNA sequencing and reverse-phase protein arrays) performed on the same set of samples and provides comprehensive molecular portraits of several subtypes of breast cancers (26). We previously reported that high activities of let-7d and miR-18a were associated with better survival outcomes ER−/HER2− breast cancer (10). However, the majority of breast tumors in the United States are hormone receptor positive. We previously showed that the miRNA-mRNA correlation structure for ER+ and ER-/HER2- is markedly different, suggesting miRNA regulatory mechanisms are cancer-subtype specific (10).
We subsequently evaluated relationships between miRNA and mRNAs expression levels in ER+ breast cancer using public databases. For each miRNA-mRNA pair, we calculated Pearson correlations between miRNA and mRNA expression levels. We inferred miRNA activity based on ActMiR method that we developed (10). miR-500a emerged as a key prognostic miRNAs for ER+ breast cancer. In the TCGA dataset, high “activity” of miR-500a was strongly associated with increased breast cancer mortality (Fig. 1A); a similar association was observed using data from Dvinge et al. (14). It is worth noting that in comparison with the “activity”, the “expression” level of miR-500a showed no associations with breast cancer survival (Fig. 1B).
Fig. 1.
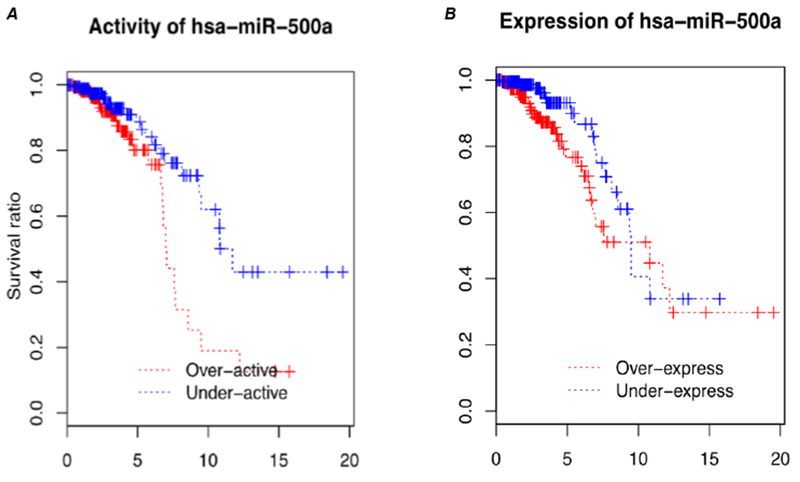
Survival curves associated with inferred activity (A) and expression levels (B) of miR-500a-5p, based on published datasets of TCGA.
Expression of miR-500a associated genes and breast cancer mortality
Given that the activity of miR-500a can only be inferred by its associated genes, we profiled 195 candidate genes (161 were miR-500a associated genes) in tumors from 606 LIBCSP patients. As shown in Table 1, among the profiled patients, 70% are postmenopausal, 20% have first degree relatives with breast cancer, and 75% and 62% are ER+ and PR+, respectively; these distributions are all similar to the overall patient cohort (11). The only notable difference is that there were fewer in situ (7% vs. 16%) and small (≤2 cm) tumors (72% vs. 80%) represented in the profiled patients compared to the overall cohort, likely due to the limited RNA yield in small and in situ tumors.
Table 1.
Demographic and clinical characteristics of profiled patients in comparison to the parent LIBCSP population (data from the parent study have been previously published (11)) and METABRIC cohort.
| All LIBCSP cases (n=1508) | Profiled LIBCSP cases (n=606) | METABRIC (n=1980) | |||||
|---|---|---|---|---|---|---|---|
| Title | Group | No | % | No | % | No | % |
| Age of diagnosis | <50 years | 407 | 27% | 158 | 26% | 424 | 21% |
| 50+ years | 1101 | 73% | 448 | 74% | 1556 | 79% | |
| Missing | 0 | 0 | 0 | ||||
| Family history of breast cancer | No first degree | 1166 | 80% | 468 | 80% | ||
| First degree | 295 | 20% | 114 | 20% | |||
| Missing | 47 | 24 | |||||
| Menopausal status | Premenopausal | 472 | 32% | 180 | 30% | 424 | 21% |
| Postmenopausal | 1006 | 68% | 414 | 70% | 1556 | 79% | |
| Missing | 30 | 12 | 0 | ||||
| Age at menarche | <12 years | 391 | 26% | 157 | 26% | ||
| 12+ years | 1104 | 74% | 442 | 74% | |||
| Missing | 13 | 7 | |||||
| Parity status | Nulliparous | 198 | 13% | 83 | 14% | ||
| Parous | 1310 | 87% | 523 | 86% | |||
| Missing | 0 | 0 | |||||
| Lactation (among parous) | Never | 841 | 64% | 332 | 63% | ||
| Ever | 469 | 36% | 191 | 37% | |||
| Missing | 0 | 0 | |||||
| Body Mass Index at reference | <22.3 | 341 | 23% | 128 | 21% | ||
| 22.3-25.1 | 367 | 25% | 151 | 25% | |||
| 25.2-29.2 | 391 | 26% | 154 | 26% | |||
| >29.2 | 392 | 26% | 169 | 28% | |||
| Missing | 17 | 4 | |||||
| Stage of breast cancer | In situ | 235 | 16% | 44 | 7% | 0 | 0% |
| Invasive | 1273 | 84% | 562 | 93% | 1980 | 100% | |
| Missing | 0 | 0 | 0 | ||||
| Tumor size | <= 2 cm | 466 | 80% | 162 | 72% | 838 | 42% |
| 2-5 cm | 102 | 18% | 56 | 25% | 1015 | 51% | |
| 5+ cm | 11 | 2% | 6 | 3% | 127 | 6% | |
| Missing | 929 | 382 | 0 | ||||
| ER status (by IHC) | Negative | 264 | 27% | 119 | 25% | 439 | 23% |
| Positive | 726 | 73% | 356 | 75% | 1498 | 77% | |
| Missing | 518 | 131 | 43 | ||||
| PR status (by IHC*) | Negative | 355 | 36% | 179 | 38% | 940 | 47% |
| Positive | 635 | 64% | 296 | 62% | 1040 | 53% | |
| Missing | 518 | 131 | 0 | ||||
| HER2 status (by IHC) | Negative | 554 | 80% | 422 | 81% | 1733 | 88% |
| Positive | 135 | 20% | 102 | 19% | 247 | 12% | |
| Missing | 819 | 82 | 0 | ||||
for PR expression status, METABRIC data had PR_status but not PR_IHC field
numbers in the table do not always add up to 1508 and 606 because of missing data
From the 195 genes profiled, 98% (n=186) were detectable in more than 50% of the samples (Fig. S3). Among the detectable genes, median raw counts varied from 10 (KAT8 probe) to 12547 (FTH1P11 probe) for candidate genes and the 6 housekeeping genes ranged from 66 (HPRT1 probe) to 1956 (GAPDH probe).
Using Cox proportional hazards models, we calculated HRs for each of miR-500a associated genes in relation to breast cancer-specific mortality, adjusted for age and tumor stage, among the LIBCSP population-based cohort of women with breast cancer. Fig. 2 displays log-transformed p-values (y-axis) and HRs (x-axis) for these associations. Detailed results are reported in Table S3. From 161 miR-500a associated genes, 74 displayed significant (FDR<0.05) associations with breast cancer-specific mortality. With increasing gene expression levels, 31 were associated with reduced breast cancer-specific mortality (HR<1), whereas 43 showed elevated risk of mortality (HR>1). Many well-known breast cancer-related genes (e.g. ESR1, AR) were among those demonstrating significant association with mortality. The strongest (by p-value) inverse association for breast cancer-specific mortality was for SUSD3 (HR=0.3, 95% CI 0.2 - 0.4, p=5.2e-10), and the strongest positive association was for BIRC5 (HR=3.3, 95% CI 2.3 - 4.8, p=5.7e-10). These 74 genes also demonstrated high degree of correlation with each other; the correlation matrix (Fig. 3) indicates two distinct expression clusters with opposing association with breast cancer mortality; such clustering was also supported by principal component analysis (Fig. S4): PC1 showed a positive association with breast cancer mortality (HR = 4.2, 95% CI 2.9-6.2, p=4.8e-14), whereas PC2 showed the opposite (HR = 0.5, 95% CI 0.4-0.8, p=5.3e-4).
Fig. 2.

Age- and stage- adjusted correlation of individual miR500a-related genes expression levels with long-term breast cancer-specific survival of LIBCSP patients. Each dot corresponds to a gene, with horizontal axis displaying hazard ratio of high expression group comparing to low expression group (i.e. genes on the left are associated with better survival, genes on the right are associated with worse survival) and vertical axis displaying logarithm of p-value of the same Cox model. Genes included in the PAM50 panel are marked with pink color.
Fig. 3.
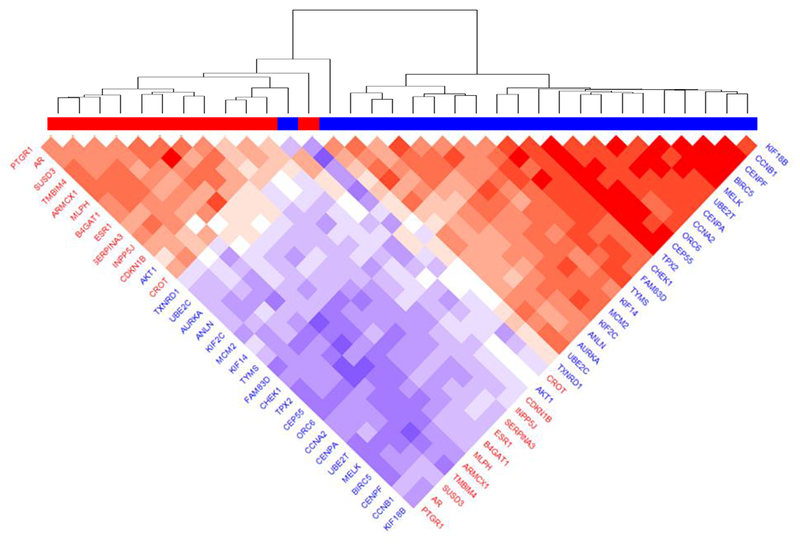
Co-expression of miR-500a-related genes with strongest association with survival, according to LIBCSP results. Blue color in gene labels and horizontal bar indicates genes associated with worse survival, red color associated with better survival. Color of the plot cells shows the correlation in respective pair of genes (red color indicating positive correlation and blue color – negative correlation).
We selected the genes to be profiled based on their association with miR-500a “activity”, both positive as well as negative associations. While a negative association suggests “direct targets” which represents the conventional mechanism of miRNA function, a positive association, which was also frequently observed, implicates mechanisms of indirect target or other modes of action. Fig. 4 depicts associations between HRs of each tested gene and its correlation with miR-500a activity by ActMiR algorithm for TCGA dataset. Based on our a priori hypothesis that high miR-500a activity confers increased breast cancer mortality, miR-500 associated genes should reside in Quadrant II or III, which is largely consistent with our findings (Fig. 4). There are a small number of genes resided in Quadrant I and IV implicating unknown modes of action. It is important to point out that 22 (including BIRC5) of the 161 miR-500a genes are also part of PAM50 gene panel (pink dots on Fig. 4); 16 showed significant association with breast cancer mortality (FDR<0.05)
Fig. 4.
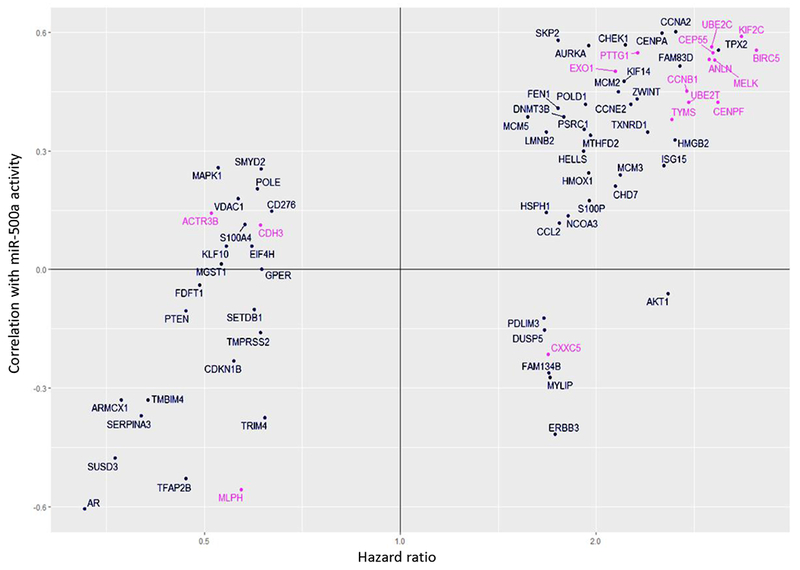
Concordance between two characteristics: association of gene expression with miR-500a activity (vertical axis: correlation coefficient) calculated by ActMiR algorithm for TCGA dataset, and association of gene expression with survival (horizontal axis: hazards ratio) calculated for Cox model for LIBCSP dataset.
Prognostic markers independent of PAM50 and ER/PR status.
Given that hormone receptor status and PAM50-derived risk scores are established factors associated with breast cancer survival, we set out to investigate whether miR-500a associated genes can predict survival, once we adjusted for these predictors.
Among the 606 tumors profiled, 78% were ER+ and 67% were PR+ resulting in 80% ER+ or PR+ (ER+/PR+) and 20% ER− and PR− (ER−PR−) based on gene expression-based categorization. These numbers are similar to what have been reported by other studies of Caucasian Americans based on IHC status (27–29) (range from 81%−86% for ER+/PR+). We first re-evaluated associations between miR-500a associated genes and breast cancer-specific mortality by adjusting for ER/PR status; 43 out of 73 genes initially associated with breast cancer mortality remained significant (FDR<0.05). When we performed stratified analyses with respect to ER/PR status, 61 genes were significantly associated with breast cancer-specific mortality in ER+/PR+ group (BIRC5 and SUSD3 remained the strongest predictors of higher and lower mortality, respectively) while only 2 genes were significant for ER−PR− group (TMPRSS2: HR=3.4, 95% CI 1.8 - 6.5; CLDN7: HR=3.3, CI 1.7 - 6.3) (Table S3).
We then investigated whether miR-500 associated genes were associated with breast cancer mortality, once we adjusted/stratified for PAM50 risk scores. All PAM50 genes were at measurable levels in all 606 LIBCSP tumor samples. Risk-Of-Recurrence (ROR) scores (high, medium and low) were computed using the algorithm (15) that takes into account expression of PAM50 genes as well as the patient’s age and tumor stage. The associations of ROR scores and breast cancer-specific mortality are shown in Fig. S5. The survival curves were similar among patients with low and medium ROR scores so we combined them as reference group (Fig. S5). As expected, high ROR was associated with increased breast cancer-specific mortality (HR=3.6, 95% CI 2.4 - 5.2, p=1.2e-10) compared to the reference group. When we evaluated ROR-adjusted HRs for all 73 genes initially associated with breast cancer mortality, 31 remained significant (FDR < 0.05). We then stratified by ROR scores, high vs. low/medium combined. In the high-risk group (249 patients, 81 deaths), 10 genes were significantly associated with breast cancer mortality (FDR<0.05), with AKT1 (HR=3.0, 95% CI 1.9-4.8) and CROT (HR=0.3, 95% CI 0.1-0.6) showing the strongest positive and negative correlations with breast cancer mortality, respectively. In the low/medium risk group (357 participants, 38 death), TMPRSS2 (HR=4.0, 95% CI 2.1-7.8) and SUSD3 (HR=0.3, 95% CI: 0.1-0.5) demonstrated significant associations with survival.
Additional evaluation of the predictive value of miR-500a-related genes was performed using a calculation of receiving operator characteristics (ROC analysis.). One of the advantages of this approach is that it is not based on the “optimal threshold” but takes the whole range of predictor’s values. As a reference, we first calculated AUC (area under curve) for the risk-of-recurrence score. Without adjustment for the survival length, AUC for the PAM-50 derived ROR-P score was very similar to the AUC of our most significantly associated gene SUSD3 (0.70 vs 0.69, Fig. 5A). However when we applied time-dependent modeling (25) using survivalROC package to consider survival length (within one year after diagnosis), SUSD3 alone outperformed ROR-P score (AUC 0.86 vs 0.62, Fig. 5B).
Fig. 5.
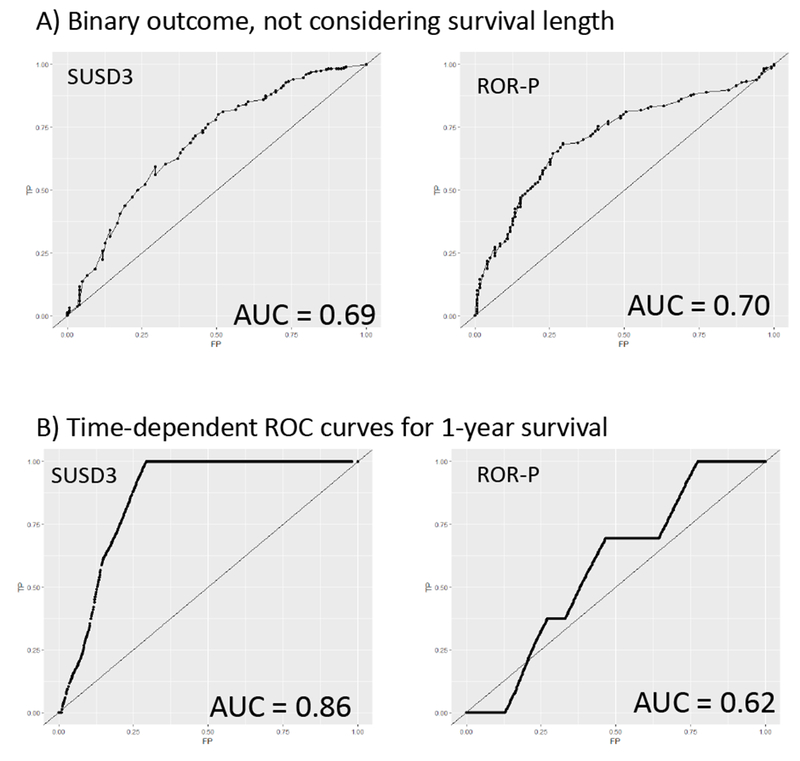
ROC curves for the SUSD3 expression level (left) and risk-of-recurrence score (right). (A) Survival as a binary outcome without adjustment for the length of survival. (B) Survival within one year time window.
Lastly, we validated our findings in two public datasets, i.e. METABRIC dataset (30) which includes 1980 breast cancer cases (77% ER+), and a combined dataset of KMPlot service kmplot.com (31) with 2862 cases (72% ER+). Most significant survival-associated genes from our study displayed the same direction of survival association in those datasets. (Fig. S6, Fig. S7). As an example, Fig. S8 shows data for our top gene SUSD3: it is consistent among all 3 datasets, and for both ER+ and ER− subsets.
Discussion
Despite the fact that overall breast cancer mortality rates declined 36% from 1989 to 2012 (2015-2016 Report from American Cancer Society), breast cancer remains the second leading cause of cancer death among US women. The decline in breast cancer mortality has been partially attributed to improvements of treatment, in which molecular phenotyping has played a significant role. Hormone receptor-positive breast cancer represents more than 80% of patients among US women (32). This category of patients is typically treated with adjuvant endocrine therapy, with or without adjuvant chemotherapy (4). Whether the patient should receive chemotherapy is a difficult choice between risk of recurrence and adverse effects of the therapy: while treatment is usually toxic and decreases patients’ quality-of-life, it is often used to prevent relapse of the tumor. Early approaches of estimating a high-risk subgroup were based on “clinical” characteristics of disease including tumor size, presence/number of lymph node metastases, and histology of tumor tissue (33), then came the inclusion of immunohistological staining for hormone receptor proteins (estrogen and progesterone receptors) (33). The simplest assay, IHC4, is based on just four immunohistochemical markers (ER, PR, HER2 and Ki67) determined in an FFPE tumor tissue sample, and was shown to have good performance in a centralized study when combined with clinical characteristics (34). However, usage of this assay in decentralized conditions is much less robust (33). All IHC-based assays share similar technical limitations: staining is highly dependent on the quality of sample, variability of antibodies used, and subjective nature of scoring of the intensity of staining. Gene expression-based assays minimize these limitations and allow measurement of multiple targets in one assay. As a result, almost all recently developed assays are based on measuring mRNA instead of protein levels. Currently there are several approved gene expression panels for clinical use, three of which have the majority of the market: 21-gene Oncotype DX™, 50-gene Prosigna™ and 70-gene Mammaprint™ (35).
MicroRNAs represent a special type of molecular mechanism of gene regulation, and have the capacity to repress almost any gene having a corresponding target sequence. Their involvement in cancer-related pathways has been clearly demonstrated, and including miRNA-related scores in disease prediction panels is a logical next step (7). It should be noted, however, that not all platforms designed for mRNA level detection are compatible with miRNA measurement, mainly due to shorter length of miRNAs. More importantly, miRNA “expression” level is not equivalent to its regulatory “activity” (8, 36), because activities of miRNAs are modulated through the accessibility of proteins or RNAs that is essential for miRNA functions and the relative abundance of miRNA to its target genes (37–39). Thus, miRNA expression levels might not be the best functional indicator. For our cases, miR-500a expression levels were measured by qPCR on a small subset of paired tumor/adjacent premalignant tissues and no significant difference was observed (Fig. S9); therefore we did not measure miR-500a expression in the entire cohort.
In our study, we reported that miR-500a activity (but not expression) was associated with survival of ER-positive patients with early stage breast cancer in two public databases. We profiled a set of miR-500a associated genes, including potential direct and indirect targets, and aimed to investigate their prognostic significance in the well-characterized population-based cohort of breast cancer patients with long-term follow-up. Our study revealed a set of genes strongly associated with the survival of breast cancer patients, in agreement with what is predicted by ActMiR (10). Moreover, most of these associations were validated in two independent populations, including a large dataset of METABRIC (30) and the combined dataset provided by KMplot.com portal (31). At the same time, gene expression data from the set of clinical samples used in this study are not sufficient to claim that ActMiR-calculated `miR-500a activity` matches real biological activity: more specific in vitro experiments need to be performed in order to justify that type of equivalence. However, the direction of associations observed in this study was largely consistent with what was predicted by ActMiR (see Fig. 4) which strongly suggests that the panel of genes we assayed was indeed related to miR-500a activity.
Although many of the predictor genes revealed in our study are novel for breast cancer prognosis, it does not mean they are better predictors of breast cancer survival because they can be highly correlated with established prognostic markers. To address this issue, we re-iterated survival analysis by taking into account two well-established prognostic markers (ER/PR status and PAM50-derived ROR score). Large proportion of genes that showed significant associations in overall breast cancer patients remained significant, even after adjusting for ER/PR status (43 out of 73) or PAM50-derived ROR scores (31 out of 73). Among the high-risk (ER−PR− or high ROR) or low-risk (ER+/PR+, or low/medium ROR) group, we were able to identify a set of genes that show significant associations with survival and differentiate the group with different survival profiles. This is an important finding because it might potentially help to better classify patients and improve precision of treatment or general care for these patients.
Although miR-500a has been identified as one of key players in breast cancer mortality, its exact role remains unknown. TargetScan (40) predicts that as many as 3816 possible target transcripts for this microRNA but none of them are experimentally validated. There is only a handful of publications which suggest that on miR-500a is a potential circulating biomarker of such pathologies as autism (41), lung cancer (42), endometriosis (43), and hepatocellular carcinoma (44).
Our study discovered a large number of miR-500a genes that demonstrated strong associations with breast cancer survival. Many of these associated with poor survival in our population-based cohort of women with breast cancer are already part of PAM50 panel (such as BIRC5, CEP55, CENPF – all known to be markers of poor survival). However, there were many others where we are the first to be report that they are predictive of breast cancer survival, even once we adjusted for the PAM50 ROR score. For example, high expression of SUSD3 (Sushi Domain-Containing Protein 3) displayed significantly reduced mortality for both HR-positive and HR-negative breast cancer cases in our population, and this finding was validated in both the METABRIC and combined KMplot datasets (Fig. S6). Its association remained significant after adjustment for ROR (HR 0.4, 95% CI 0.3 - 0.7). More importantly, SUSD3 level effectively stratified low-ROR group (HR 0.3, 95% CI 0.1 - 0.5) identifying the patients with poor survival within this group. According to AUC values, in our study the SUSD3 alone performed equaly well with ROR score in its predictability of breast cancer survival and performed even better when adjusted for survival length within one-year window (Fig. 5). This gene was shown to be a survival biomarker based on METABRIC data (45), and increased expression of corresponding protein was reported in breast cancer tumor tissue (46). At the same time, there was only one previous study focused on deciphering molecular mechanism of this gene. Moy et al. (47) showed that SUSD3 was a direct transcriptional target of estrogen receptor in MCF7 breast cancer cells, and its expression was induced after stimulation with estradiol. SUSD3 product was shown to be localized at plasma membrane, and its knockdown lead to decreased cell motility and proliferation (47). These results cannot easily fit with the inverse association with breast cancer mortality in our study. The molecular mechanism of SUSD3 deserves to be further investigated experimentally. Another gene highly correlated with breast cancer survival after adjustment for PAM50-derived ROR scores (HR 0.4, 95% CI 0.2 - 0.6) was CROT (Carnitine O-Octanoyltransferase). Again, its association was highly significant not only in our dataset but also by KMplot data (METABRIC data not available for CROT: gene symbol not found in the list). This gene is known to be involved in lipid acids metabolism, but was not previously linked to cancer. Similar inverse association was shown for ARMCX1 (armadillo repeat containing, X-linked 1) gene in patients from all three datasets, including ROR-adjusted LIBCSP patients (HR 0.5, 95% CI 0.4 - 0.8), with other studies having confirmed it as potential tumor suppressor gene (48).
While our study has many strength including a well-characterized population-based cohort with long follow-up coupled with a novel systems biology approach, we acknowledge limitations of the study. Part of those limitations come from the lack of detailed clinical information. For example, we did not have integrated staging information (I – IIA – IIB etc.), and only used invasiveness status as an available stage. In addition, we did not perform in-depth analysis on treatment, which may influence breast cancer survival. Such exclusion stems from several reasons. First, only ~66% participants completed the follow-up survey, which may introduce bias to the cohort. Second, only information on the first course of treatment was collected while future therapy could be different depending on disease progression. Third, multi-center nature of this study (more than 30 participating hospitals) assumes there could be certain heterogeneity in choice of the treatment strategies and the way it was reported. Finally and importantly, by definition, a confounder (i.e. treatment) has to be related to both the independent variable (i.e. gene expression) and outcome (i.e. survival). In our case, all tumor samples were collected prior to treatment other than surgery, the treatment and molecular profile of the tumor should be considered independent. Also, treatment is part of the causal pathway (being impacted by tumor stage or hormone receptor status at diagnosis) and including a causal intermediate in the model can bias the effect estimates (49).
In summary, by leveraging public databases and our own epidemiologic studies and cooperating systems biology approach, we identified a key microRNA, miR-500a, that appeared to be strongly linked with breast cancer mortality across platforms. More importantly, we identified a miR-500a associated gene panel that was not only strongly associated with breast cancer mortality but also can differentiate survival profiles with existing prognostic factors such as hormone receptor status and PAM50 derived risk of recurrence. These results suggest miR-500a associated genes may improve prognostic efficiency of current molecular phenotyping and ultimately improve breast cancer survival. Our study on these genes sheds light on breast cancer progression mechanisms and may lead to development of new targets for cancer therapy.
Supplementary Material
Translational Relevance.
Despite recent advances in molecular phenotyping of breast cancer which have significantly advanced our capabilities to treat breast cancer and improve survival, breast cancer still is the second leading cause of cancer death in American women; identifying additional molecular markers that can refine the risk prediction for long-term survival is urgently needed. Combining systems biology and epidemiology, we identified a key microRNA, miR-500a, that played an important role in breast cancer mortality. Importantly, we identified a miR-500a associated gene panel that was strongly associated with breast cancer-specific mortality in a population-based cohort of women with breast cancer, and this panel is independent of existing prognostic factors such as hormone receptor status and PAM50-derived risk of recurrence. Results were validated in independent datasets. These results suggest miR-500a associated gene panel may improve prognostic efficiency of current molecular phenotyping and ultimately improve breast cancer survival.
ACKNOWLEDGEMENTS
This work was supported by grant from the National Institutes of Health (NIH R01 CA172460) and in part by grants (5U01ES019459, UO1CA/ES66572, UO1CA66572).
Footnotes
Disclosure of Potential Conflicts of Interest:
The authors have no competing financial interests to declare
References
- 1.Gingras I, Desmedt C, Ignatiadis M, Sotiriou C. CCR 20th Anniversary Commentary: Gene-Expression Signature in Breast Cancer--Where Did It Start and Where Are We Now? Clinical cancer research : an official journal of the American Association for Cancer Research. 2015. November 01;21(21):4743–6. [DOI] [PubMed] [Google Scholar]
- 2.Hyams DM, Schuur E, Angel Aristizabal J, Bargallo Rocha JE, Cabello C, Elizalde R, et al. Selecting postoperative adjuvant systemic therapy for early stage breast cancer: A critical assessment of commercially available gene expression assays. Journal of surgical oncology. 2017. February 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith IE, Yeo B, Schiavon G. The optimal duration and selection of adjuvant endocrine therapy for breast cancer: how long is enough? American Society of Clinical Oncology educational book American Society of Clinical Oncology Meeting 2014:e16–24. [DOI] [PubMed] [Google Scholar]
- 4.National Comprehensive Cancer Network. NCCN Guidelines. Breast Cancer (Version 2.2016). [cited 2017 Feb 6]; Available from: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. [Google Scholar]
- 5.Prat A, Lluch A, Turnbull AK, Dunbier AK, Calvo L, Albanell J, et al. A PAM50-Based Chemoendocrine Score for Hormone Receptor-Positive Breast Cancer with an Intermediate Risk of Relapse. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016. November 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bahrami A, Aledavood A, Anvari K, Hassanian SM. The prognostic and therapeutic application of microRNAs in breast cancer: Tissue and circulating microRNAs. 2017. January 21. [DOI] [PubMed] [Google Scholar]
- 7.Haakensen VD, Nygaard V, Greger L, Aure MR, Fromm B, Bukholm IR, et al. Subtype-specific micro-RNA expression signatures in breast cancer progression. International journal of cancer. 2016. September 01;139(5):1117–28. [DOI] [PubMed] [Google Scholar]
- 8.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, et al. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nature methods. 2012. July 01;9(8):840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nature methods. 2007. September;4(9):721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee E, Ito K, Zhao Y, Schadt EE, Irie HY, Zhu J. Inferred miRNA activity identifies miRNA-mediated regulatory networks underlying multiple cancers. Bioinformatics. 2016. January 1;32(1):96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gammon MD, Neugut AI, Santella RM, Teitelbaum SL, Britton JA, Terry MB, et al. The Long Island Breast Cancer Study Project: description of a multi-institutional collaboration to identify environmental risk factors for breast cancer. Breast cancer research and treatment. 2002. June;74(3):235–54. [DOI] [PubMed] [Google Scholar]
- 12.Cleveland RJ, Eng SM, Abrahamson PE, Britton JA, Teitelbaum SL, Neugut AI, et al. Weight gain prior to diagnosis and survival from breast cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007. September;16(9):1803–11. [DOI] [PubMed] [Google Scholar]
- 13.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007. July 6;27(1):91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dvinge H, Git A, Graf S, Salmon-Divon M, Curtis C, Sottoriva A, et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature. 2013. May 16;497(7449):378–82. [DOI] [PubMed] [Google Scholar]
- 15.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009. March 10;27(8):1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossner P Jr., Gammon MD, Zhang YJ, Terry MB, Hibshoosh H, Memeo L, et al. Mutations in p53, p53 protein overexpression and breast cancer survival. Journal of cellular and molecular medicine. 2009. September;13(9b):3847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stacklies W, Redestig H, Scholz M, Walther D, Selbig J. pcaMethods--a bioconductor package providing PCA methods for incomplete data. Bioinformatics (Oxford, England). 2007. May 01;23(9):1164–7. [DOI] [PubMed] [Google Scholar]
- 18.Harris LN, Ismaila N, McShane LM, Andre F, Collyar DE, Gonzalez-Angulo AM, et al. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women With Early-Stage Invasive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016. April 01;34(10):1134–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox DR. Regression Models and Life-Tables. Journal of the Royal Statistical Society Series B (Methodological). 1972;34(2):187–220. [Google Scholar]
- 20.Borgan Ø Modeling Survival Data: Extending the Cox Model Therneau Terry M. and Grambsch Patricia M., Springer-Verlag, New York, 2000. No. of pages: xiii + 350. Price: $69.95. ISBN 0-387-98784-3. Statistics in Medicine. 2001;20(13):2053-4. [Google Scholar]
- 21.Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA, et al. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast cancer research and treatment. 2013. July;140(2):219–32. [DOI] [PubMed] [Google Scholar]
- 22.Rothman KJ, Greenland S, Lash TL. Modern epidemiology. 3rd ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008. x, 758 p. p. [Google Scholar]
- 23.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association. 1958;53(282):457–81. [Google Scholar]
- 24.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics. 2011. March 17;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000. June;56(2):337–44. [DOI] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012. October 4;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA: a cancer journal for clinicians. 2016. Jan-Feb;66(1):31–42. [DOI] [PubMed] [Google Scholar]
- 28.Baquet CR, Mishra SI, Commiskey P, Ellison GL, DeShields M. Breast cancer epidemiology in blacks and whites: disparities in incidence, mortality, survival rates and histology. Journal of the National Medical Association. 2008. May;100(5):480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joslyn SA. Hormone receptors in breast cancer: racial differences in distribution and survival. Breast cancer research and treatment. 2002. May;73(1):45–59. [DOI] [PubMed] [Google Scholar]
- 30.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012. April 18;486(7403):346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment. 2010. October;123(3):725–31. [DOI] [PubMed] [Google Scholar]
- 32.Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LA, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. Journal of the National Cancer Institute. 2014. April 28;106(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harbeck N, Sotlar K, Wuerstlein R, Doisneau-Sixou S. Molecular and protein markers for clinical decision making in breast cancer: today and tomorrow. Cancer treatment reviews. 2014. April;40(3):434–44. [DOI] [PubMed] [Google Scholar]
- 34.Barton S, Zabaglo L, A’Hern R, Turner N, Ferguson T, O’Neill S, et al. Assessment of the contribution of the IHC4+C score to decision making in clinical practice in early breast cancer. British journal of cancer. 2012. May 22;106(11):1760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markopoulos C, van de Velde C, Zarca D, Ozmen V, Masetti R. Clinical evidence supporting genomic tests in early breast cancer: Do all genomic tests provide the same information? European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2016. August 31. [DOI] [PubMed] [Google Scholar]
- 36.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, et al. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012. August;9(8):840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arvey A, Larsson E, Sander C, Leslie CS, Marks DS. Target mRNA abundance dilutes microRNA and siRNA activity. Mol Syst Biol 2010. April 20;6:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010. June 24;465(7301):1033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010. September;11(9):597–610. [DOI] [PubMed] [Google Scholar]
- 40.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015. August 12;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kichukova TM, Popov NT, Ivanov IS, Vachev TI. Profiling of Circulating Serum MicroRNAs in Children with Autism Spectrum Disorder using Stem-loop qRT-PCR Assay. Folia medica. 2017. March 01;59(1):43–52. [DOI] [PubMed] [Google Scholar]
- 42.Joerger M, Baty F, Fruh M, Droege C, Stahel RA, Betticher DC, et al. Circulating microRNA profiling in patients with advanced non-squamous NSCLC receiving bevacizumab/erlotinib followed by platinum-based chemotherapy at progression (SAKK 19/05). Lung cancer (Amsterdam, Netherlands). 2014. August;85(2):306–13. [DOI] [PubMed] [Google Scholar]
- 43.Cosar E, Mamillapalli R, Ersoy GS, Cho S, Seifer B, Taylor HS. Serum microRNAs as diagnostic markers of endometriosis: a comprehensive array-based analysis. Fertility and sterility. 2016. August;106(2):402–9. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z, Ge S, Wang X, Yuan Q, Yan Q, Ye H, et al. Serum miR-483-5p as a potential biomarker to detect hepatocellular carcinoma. Hepatology international. 2013. March;7(1):199–207. [DOI] [PubMed] [Google Scholar]
- 45.Ou Yang TH, Cheng WY, Zheng T, Maurer MA, Anastassiou D. Breast cancer prognostic biomarker using attractor metagenes and the FGD3-SUSD3 metagene. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2014. December;23(12):2850–6. [DOI] [PubMed] [Google Scholar]
- 46.Zhao S, Chen SS, Gu Y, Jiang EZ, Yu ZH. Expression and Clinical Significance of Sushi Domain- Containing Protein 3 (SUSD3) and Insulin-like Growth Factor-I Receptor (IGF-IR) in Breast Cancer. Asian Pacific journal of cancer prevention : APJCP. 2015;16(18):8633–6. [DOI] [PubMed] [Google Scholar]
- 47.Moy I, Todorovic V, Dubash AD, Coon JS, Parker JB, Buranapramest M, et al. Estrogen-dependent sushi domain containing 3 regulates cytoskeleton organization and migration in breast cancer cells. Oncogene. 2015. January 15;34(3):323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao Y, Wu JY, Zeng F, Liu GL, Zhang HT, Yun H, et al. ALEX1 Regulates Proliferation and Apoptosis in Breast Cancer Cells. Asian Pacific journal of cancer prevention : APJCP. 2015;16(8):3293–9. [DOI] [PubMed] [Google Scholar]
- 49.Rothman KJ. Epidemiology : an introduction. 2nd ed. New York, NY: Oxford University Press; 2012. viii, 268 p. p. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.