

Abstract
Multidrug-resistant Vibrio parahaemolyticus has become a significant public health concern. The development of effective drugs and vaccines against Vibrio parahaemolyticus is the current research priority. Thus, we aimed to find out effective drug and vaccine targets using a comprehensive genome-based analysis. A total of 4822 proteins were screened from V. parahaemolyticus proteome. Among 16 novel cytoplasmic proteins, ‘VIBPA Type II secretion system protein L’ and ‘VIBPA Putative fimbrial protein Z’ were subjected to molecular docking with 350 human metabolites, which revealed that Eliglustat, Simvastatin and Hydroxocobalamin were the top drug molecules considering free binding energy. On the contrary, ‘Sensor histidine protein kinase UhpB’ and ‘Flagellar hook-associated protein of 25 novel membrane proteins were subjected to T-cell and B-cell epitope prediction, antigenicity testing, transmembrane topology screening, allergenicity and toxicity assessment, population coverage analysis and molecular docking analysis to generate the most immunogenic epitopes. Three subunit vaccines were constructed by the combination of highly antigenic epitopes along with suitable adjuvant, PADRE sequence and linkers. The designed vaccine constructs (V1, V2, V3) were analyzed by their physiochemical properties and molecular docking with MHC molecules- results suggested that the V1 is superior. Besides, the binding affinity of human TLR-1/2 heterodimer and construct V1 could be biologically significant in the development of the vaccine repertoire. The vaccine-receptor complex exhibited deformability at a minimum level that also strengthened our prediction. The optimized codons of the designed construct was cloned into pET28a(+) vector of E. coli strain K12. However, the predicted drug molecules and vaccine constructs could be further studied using model animals to combat V. parahaemolyticus associated infections.
Introduction
Vibrio parahaemolyticus, a highly reported pathogenic bacteria of aquatic environment, has emerged as the leading cause of seafood-associated gastroenteritis and a significanthazardfor global aquaculture [1–3]. The overgrowing population, with increased purchasing power worldwide has enhanced the demand for and export potential of seafood, resulting in the steady expansion of the aquaculture industry [4]. However, the sector has continuously been challenged by aquatic animal health problems, which is a significant constraint to the development of this sector [5]. Besides, Multiple drug resistance (MDR) has been recognized as an essential global threat issue to food safety [6]. The continuous and inappropriate use of antibiotics in the aquaculture industry favors the development of a variety of resistant isolates and the dissemination of resistance genes within the bacterial population [7]. V. parahaemolyticus has been reported to show multidrug resistance during aquaculture production [8], which raised the concern about public health and economic threat of this bacterium [9].
Though V. parahaemolyticus was first isolated in 1952, reports demonstrated the recent outbreaks of V. parahaemolyticus are more severe [10,11]. On the recent outbreak in the city of Osaka (Japan), acute gastroenteritis was reported in 272 individuals, 20 of whom died [12]. To date, V. parahaemolyticus has been responsible for 20–30% of food poisoning cases in Japan and sea foodborne diseases in many Asian countries [13]. A total 802 outbreaks of food-borne diseases have been reported in 13 of the coastal provinces of eastern China, causing more than 17,000 individuals to become ill [14], where V. parahaemolyticus attributed the most significant number (40.1%) of these cases [15,16]. The leading cause of human gastroenteritis associated with seafood consumption in the United States is V. parahaemolyticus [17]. Centers for Disease Control and Prevention (CDC) declared it as a significant foodborne bacterium compared to other Vibrio species, which was responsible for approximately 34,664 foodborne cases annually in the USA [18].
The food poisoning caused by V. parahaemolyticus usually occurs in summer and is predominantly associated with different kinds of seafood, including crab, shrimp, shellfish, lobster, fish and oysters [19,20]. V. parahaemolyticus is usually found in a free-swimming state, with its motility conferred by a single polar flagellum affixed to inert and animate surfaces including zooplankton, fish, shellfish or any suspended matter underwater [21]. Among the whole range of seafood, shellfish is regarded as a high-risk food because it is infested with large populations of bacteria, including V. parahaemolyticus [22]. Illness is inevitable, once consumers eat undercooked contaminated seafood [23]. The symptoms of the disease include diarrhea, vomiting, abdominal pain, nausea and low-grade fever. In most cases, the disease is self-resolving. However, V. parahaemolyticus may cause a more debilitating and dysenteric form of gastroenteritis [24]. Uncommonly, in immunocompromised patients, it may progress into a life-threatening fulminant necrotizing fasciitis characterized by rapid necrosis of subcutaneous tissue [25]. In rare cases, V. parahaemolyticus causes septicemia, which is also associated with a high mortality rate [26]. Also, V. parahaemolyticus is one of the major pathogens of cultured mud crabs and cause acute hepatopancreaticnecrosis disease (AHPND) in shrimp [27]. Usually, 99% of clinical V. parahaemolyticus isolates are known to be pathogenic, whereas the majority of the environmental isolates are non-pathogenic [28]. Nonetheless, around 0–6% of the environmental isolates are identified as pathogenic carrying virulence genes [3]. During infection, V. parahaemolyticus uses the adhesion factors to bind to the fibronectin and phosphatidic acid on the host cell, thus releasing different effectors and toxins into the cytoplasm, causing cytotoxicity and serious diseases [21]. Thermostable direct hemolysin (TDH) and TDH-related hemolysin (TRH) are considered two major virulence factors of this pathogen due to the invasiveness and roles in disease pathogenesis [3]. TDH, which is prevalent in 95% of clinical V. parahaemolyticus isolates, can lyse red blood cells when secreted [29]. Whole-genome sequencing of V. parahaemolyticus confirmed that the pathogen possesses two sets of type III secretion system (i.e. T3SS1 and T3SS2) genes [30], where T3SS1 is involved in cytotoxity and T3SS2 is responsible for enterotoxicity [31].
Many antibiotics are no longer effective in hospitals to treat V. Parahaemolyticus infections [32,33]. First-generation antibiotics, including ampicillin are extensively used in aquaculture resulting in reduced susceptibility andlow efficacy of ampicillin for Vibrio sp. treatment [34]. Literature also reported higher resistance to third-generation antibiotics such as cephalosporin, cefotaxime, carbapenemsand ceftazidime by V. parahaemolyticus isolates [20,35] which enhanced the necessity of searching safe and more effective drugs for combating infections caused by V. parahaemolyticus in the future. However, the development of new antibiotics is difficult and time-consuming. Recent progress in the field of computational biology and bioinformatics has generated various in silico analysis and drug designing approaches. Thus eliminating the time and cost involved in the early trial phase before going into the drug development phase [36]. Subtractive proteomics is one such in silico strategy that helps to facilitate the selection, processing, and development of strain-specific drugs against various pathogens [37]. It can be utilized to identify drug targets based on the determination of essential and nonhomologous proteins within the pathogenic organism [38,39]. The term ‘Druggability’ is used to describe a biological target (e.g. protein) with the potential to bind with high affinity to a drug [40]. The concept is often utilized in drug discovery which reflects the ability of a druggable target to be modulated by small drug-like molecules. Various novel drug targets have already been successfully identified for S. typhi meningitides sero group B using the mentioned approach [39].
Moreover, in silico docking studies between the identified drug targets and existing drugs with slight modification may lead to the discovery of novel drugs for the treatment of infections [41,42]. As a result, a wide range of drug targets and lead compounds can be identified before laboratory experimentation to save time and money. The study was designed to employ a comprehensive genome-based analysis of Vibrio parahaemolyticus for identifying novel therapeutic targets as well as suitable drugs and vaccine molecules through subtractive proteomics and vaccinomics approaches.
Materials and methods
The whole proteome of V. parahemolyticus was analyzed according to subtractive proteomics approach to recognize novel drug targets as well as vaccine candidates. The overall workflow for subtractive proteomic analysis and vaccinomics approach has been illustrated in results section.
Retrieval of complete proteome and identification of essential proteins
The whole proteome of V. parahemolyticusstrain was retrieved from NCBI Genome database. Paralogous sequences were excluded from the proteome of V. parahemolyticus by using CD-HIT [43]. With a cutoff score of 0.6, proteins with more than 60% identity were excluded. Remaining proteins were subjected to BLASTp against Homo sapiens human Refseq proteome in ‘Ensemble Genome Database 92’ using threshold expectation value (E- value) 10−3 as the parameter. Proteins were assumed as homologous were excluded if any significant hit above the threshold value 10−4 was found. The remaining non-homologous proteins were subjected to the Database of Essential Genes (DEG) [44]. Proteins hit with expectation value ≤10–100, identity ≥ 25% were listed for metabolic pathway analysis considering as essential non-homologous proteins of V. parahemolyticus.
Analysis of metabolic pathways
Kyoto Encyclopedia of Genes and Genomes (KEGG) which contains complete metabolic pathways present in living organisms [45]. Metabolic pathways of V. parahemolyticus were analyzed against the human metabolic pathways through the KEGG server. All metabolic pathways present in the pathogen (V. parahemolyticus) and host (H. sapiens) were collected from the KEGG PATHWAY database using three letters KEGG organism code ‘vpa’ and ‘has’ respectively. A comparison was made in order to recognize the unique metabolic pathways only present in the V. parahemolyticus, while the remaining pathways of the pathogen were grouped as a common one. Identified host non-homologous, essential proteins of V. parahemolyticus were subjected to BLASTp through the KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server at KEEG. Proteins present only in the unique metabolic pathways of the pathogen were listed for further analysis.
Druggability analysis and identification of novel drug targets
A ‘druggable’ target needs to have the potentiality to bind to the drugs and drug-like molecules with high affinity. Shortlisted unique proteins were screened through the database of DrugBank 5.1.0 [46] using default parameters to identify both druggable proteins and novel therapeutic targets.
‘Anti-target’ analysis and prediction of subcellular localization
This analysis was performed to avoid any kind of cross-reactivity, and toxic effects due to docking between the drugs administered for the pathogen and the host ‘anti-targets’.‘Anti-targets’ are gene products that show cross-reactivity with administered therapeutics. Novel drug targets were subjected to BLASTp analysis in the NCBI blast program against these human ‘anti-targets’ setting an E-value <0.005, query length >30%, identity <25% as parameters. Proteins were showing a <25% identity that was listed for subcellular localization analysis. Besides, proteins functioning in cytoplasm can be used as putative drug targets, while surface membrane proteins can be considered both as drug targets and vaccine candidates. PSORTb v3.0.2 CELLO v.2.5), ngLOC servers were used to predict subcellular localization of shortlisted pathogen-specific essential proteins.
Human microbiome non-homology analysis
Both membrane and cytoplasmic proteins were subjected to BLASTp through NCBI protein blast server against the dataset present in the Human Microbiome Project server (https://www.hmpdacc.org/hmp/) (BioProject-43021) [47] with a cutoff score 0.005. Membrane proteins showing <45% similarity were selected for vaccine candidacy, whereas cytoplasmic proteins showing <45% similarity were selected for protein-protein interaction analysis.
Analysis of Virulence Factors (VF’s) and Protein-Protein Interactions studies (PPIs)
Virulence factors are responsible for modulating or degrading host defense mechanisms by bacteria. Novel cytoplasmic proteins with the least similarity with the human microbiome were subjected to BLASTp search against the database of protein sequences from the VFDB core dataset [48] with default hit with cut-off bit score >100, and E-value was 0.0001. The protein-protein interactions studies (PPIs) of selected shortlisted proteins were predicted using STRING v10.5 [49]. PPIs with a high confidence score (≥90%) were considered to avoid false-positive results. Only characterized proteins were subjected to BLASTp.
Screening of drug molecules against novel cytoplasmic proteins
All the pharmaco-metabolites reported in the Human Metabolites Database (www.hmdb.ca) were used for the screening of suitable drugs. Molecular docking was performed against predicted drug targets (novel cytoplasmic proteins) by using AutoDock Vina tools [50]. The size of the grid box was set to 54 A° x 74 A° x 126 A° (x, y and z) and 65A° x 85 A° x 65 A° (x, y and z) with 1 A° spacing between the grid points for two cytoplasmic therapeutic target proteins (Q87TC9 and Q87165 respectively).
Screening of novel outer membrane proteins for vaccine construction
The VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/) was used for the investigation of protein immunogenicity to find the most potent antigenic outer membrane proteins [51]. Proteins were prioritized based on their antigenic score (threshold value 0.4) and sequence similarity with human microbiota.
T-cell epitope prediction, transmembrane topology screening and antigenicity analysis
MHC-I (http://tools.iedb.org/mhci/) and MHC-II prediction tool (http://tools.iedb.org/mhcii/) prediction tool of the Immune Epitope Database were used to predict the MHC-I binding and MHC-II binding peptides, respectively. To predict the transmembrane helices in proteins [52] and to determine epitope antigenicity [51], TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) and VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/) were utilized.
Population coverage, allergenicity, toxicity and conservancy analysis
Population coverage for each epitope was analyzed by the IEDB population coverage calculation tool (http://tools.iedb.org/population/) [53]. The most potent antigenic epitopes were selected and allowed for determining the allergenicity patternvia four servers named AllergenFP [54], AllerTOP (http://www.ddg-pharmfac.net/AllerTop/) [55], Allermatch (http://www.allermatch.org/allermatch.py/form) [56] and Allergen Online [57]. Moreover, the ToxinPred server predicted the toxicity level of the proposed epitopes (http://crdd.osdd.net/raghava/toxinpred/) [58]. The conservancy level determines the efficacy of epitope candidates to confer broad-spectrum immunity. For revealing the conservancy pattern, homologous sequence sets of the selected antigenic proteins were retrieved from the NCBI database by using the BLASTp tool. Further, the epitope conservancy analysis tool (http://tools.iedb.org/conservancy/) at IEDB was selected for the analysis of the conservancy pattern.
Prediction of 3D structures for superior epitopes and analysis of molecular docking
Top-ranked epitopes were subjected to the PEP-FOLD server to predict peptide structures [59]. Depending on the available structures deposited in Protein Data Bank (PDB) database, HLA-A*11:01 and HLA-DRB1*04:01 were selected for docking analysis with MHC class I and class II binding epitopes respectively. MGLTools were used to visualize and analyze the molecular structures of biological compounds [60]. The grid box was set to 28 A°, 18 A°, 20 A° (x, y and z) with a default value of 1.0 A° spacing by Autodock Vina at 1.00- °A spacing. The exhaustiveness parameter was kept at 8.00, and the number of outputs was set at 10 [60]. Output PDBQT files were converted in PDB format using Open Babel. The docking interaction was visualized with the PyMOL molecular graphics system (https://www.pymol.org/).
Identification of B-Cell epitope
B-cell epitopes were predicted for both proteins to find the potential antigens that would interact with B lymphocytes and initiate the immune response. Several tools from IEDB i.e. Kolaskar and Tongaonkar antigenicity scale [61], Karplus and Schulz flexibility prediction [62], Bepipred linear epitope prediction analysis [63], Emini surface accessibility prediction [64], Parker hydrophilicity prediction [65] and Chou and Fasman beta-turn prediction [66] were used to identify the B-cell epitopes antigenicity depending on six different algorithms.
Epitope cluster analysis and vaccine construction
Epitope cluster analysis tool from IEDB was used to identify the epitope clusters with overlapping peptides for both proteins using the top CTL, HTL and BCL epitopes as input. The identified clusters and singletons were further utilized to design construct. Vaccine sequences started with an adjuvant followed by the top CTL epitopes, top HTL epitopes and BCL epitopes respectively for both proteins. Three vaccine constructs i.e. V1, V2 and V3, each associated with different adjuvants including beta-defensin (a 45 mer peptide), L7/L12 ribosomal protein and HABA protein (Mycobacterium tuberculosis, accession number: AGV15514.1) [67] were constructed. PADRE sequence and different linkers, for instance, EAAAK, GGGS, GPGPG and KK linkers, were also used to construct effective vaccine molecules.
Allergenicity, antigenicity and solubility prediction of different vaccine constructs
The AlgPred v.2.0 [68] and AllerTOP v.2.0 [55] servers were utilized to predict the non-allergic behavior of the vaccine constructs. For proposing the superior vaccine candidate, the VaxiJen v2.0 server [51] was utilized. The probable antigenicity of the constructs was determined through an alignment-independent algorithm. Protein-sol software [69] predicted the solubility score of the proposed vaccines.
Physicochemical characterization and secondary structure analysis
The ProtParam, a tool from Expasy's server (http://expasy.org/cgi-bin/protpraram) [70,71] was used to characterize the vaccine proteins functionally–including molecular weight, isoelectric pH, aliphatic index, hydropathicity, instability index, GRAVY values and estimated half-lifeand other physicochemical properties were investigated. The PSIPRED v3.3 [72] were used to predict the alphahelix, betasheet and coil structure of the vaccine protein. The polar, nonpolar and aromatic regions were also determined.
Tertiary structure prediction, refinement, validation and disulfide engineering of vaccine construct
The I-TASER server [73] was employed for determining the 3D structure of designed vaccine constructs based on the degree of similarity between the target protein and available template structure from PDB. Refinement was performed using ModRefiner [73]. The refined protein structure was validated through the Ramachandran plot assessment by the MolProbity software [74]. Residues in the highly mobile region of the protein exhibit the potential to be mutated with cysteine. Pairs of residues with propergeometry and the ability to form a disulfide bond were detected by the DbD2 server to perform disulfide engineering [75]. The value of chi3 considered for the residue screening was between −87 to +97 while the energy considered was <2.5.
Protein-protein docking and molecular dynamics simulation
The binding affinity of the vaccine constructs with different HLA alleles and human immune receptors, ClusPro 2.0 [76], hdoc [77] and PatchDock server [78] were applied. Desirable complexes were identified according to better electrostatic interaction and free binding energy following refinement via the FireDock server [79]. The iMODS server was used to explain the collective motion of proteins via analysis of normal modes (NMA) in internal coordinates [80]. Essential dynamics is a powerful tool and alternative to the costly atomistic simulation that can be compared to the normal modes of proteins to determine their stability [81]. The server predicted the direction and magnitude of the immanent motions of the complex in terms of deformability, eigenvalues, B-factors and covariance. The structural dynamics of the protein-protein complex was investigated [82].
Codon adaptation, in silico cloning and similarity analysis with human proteins
A codon adaptation tool (JCAT) was used to adapt the codon usage to the well-characterized prokaryotic organisms for accelerating the expression rate in it. Rho-independent transcription termination, prokaryote ribosome binding site and cleavage sites of restriction enzyme ApaI and BglI were avoided while using the server [83]. The optimized sequence of vaccine protein V1 was reversed, followed by conjugation with ApaI and BglI restriction site at the N-terminal and C-terminal sites, respectively. SnapGene [84] restriction cloning module was used to insert the adapted sequence into the pET28a(+) vector between ApaI (1334) and BglI (2452). At last, human sequence similarity analysis of the proposed vaccine with human proteins was done by using NCBI protein-protein Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi), and here blast was done against Homo sapiens (taxid: 9606) dataset.
Results
Various bioinformatics tools and databases were used to analyze the entire proteome of V. parahemolyticus through subtractive proteomics and vaccinomics approach. The step by step results (or workflow in Figs 1 and 2) from the complete computational analysis was presented in Table 1.
Fig 1. Proteome exploration of Vibrio parahaemolyticus to identify novel drug targets.
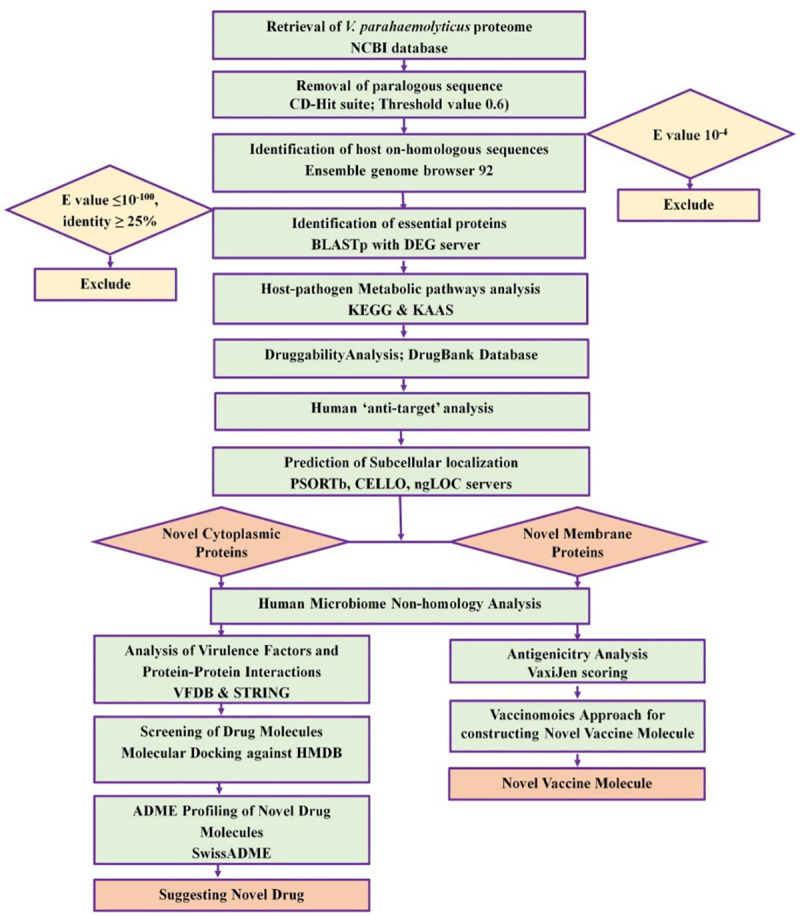
Fig 2. Flow chart summarizing the protocols over multi-epitope subunit vaccine development against V. parahaemolyticus through reverse vaccinology approach.
Table 1. Subtractive proteomics analysis scheme.
| Sl. No. | Steps | No. of proteins | Protein sets |
|---|---|---|---|
| 1 | Total number of proteins | 4822 | Set 0 (S1 File) |
| 2 | Non-paralogous (>60% identical) in CD-Hit | 4729 | Set 1 (S2 File) |
| 3 | proteins with >100 amino acids | 4123 | Set 2 (S3 File) |
| 4 | Number of proteins nonhomologous to H. sapiens using BLASTp (E value 10−3) | 3164 | Set 3 (S4 File) |
| 5 | Essential proteins in DEG 15.2 server (E value ≤10−100, Bit score >100) | 1107 | Set 4 (S5 File) |
| 6 | Essential Proteins involved only in unique metabolic pathways (KAAS at KEGG) | 96 | Set 5 (S9 File) |
| 7 | Essential proteins found to be novel in DrugBank 5.1.0 (using default parameters) | 41 | Set 6 (S10 File) |
| 8 | Novel drug target proteins non-homologous to ‘anti-targets’ using BLASTp (E value <0.005, Identity <25%, Query length >30%) | 41 | Set 7 (S11 File) |
| 9 | Essential cytoplasmic proteins using PSORTb, CELLO, ngLOC, PSLpred | 16 | Set 8 (S12 File) |
| 10 | Proteins showing <45% similarity with human microflora proteins | 9 | Set 9 (S13 File) |
| 11 | Identified virulence associated novel proteins by VFDB analysis | 2 | Set 10 (Table 4) |
| 12 | Essential membrane proteins using PSORTb, CELLO, ngLOC, PSLpred | 25 | Set 11 (S14 File) |
| 13 | Identified vaccine targets having less similarities with human microflora proteins and antigenicity | 2 | Set 12 (Table 7) |
Retrieval of complete proteome and identification of essential proteins
The whole proteome of V. parahemolyticus strain O3:K6 was extracted from NCBI database (S1 File) containing 4822 proteins (Set 0). Paralogous protein sequences of the pathogen. A total of 93 paralogous sequence above >60% similarity were identified through the CD-hit server and removed, leaving 4729 non-paralogous protein sequences in Set 1 (S2 File). Among these proteins, proteins with >100 residues (4123 proteins) (Set 2) were only considered (S3 File) for further analysis. Again, proteins showing significant similarity with human RefSeq proteins (1143 proteins) were excluded from the list designated as Set 3 (S4 File). Analysis of remaining proteins throughthe DEG server revealed only proteins that are essential (Set 4) for the survival of the pathogen (S5 File).
Analysis of metabolic pathways
The KEGG server contains 131 metabolic pathways for V. parahaemolyticus (S6 File) and 325 pathways for humans (S7 File). Through manual comparison, 40 metabolic pathways were found to be pathogen-specific and are provided in S8 File. Proteins involved in these unique pathways can be selected as drug targets. Non-homologous essential proteins subjected to BLASTp in the KAAS server at KEGG revealed that 96 proteins among 1107 assigned both KO (KEGG Orthology) and metabolic pathways that further deputed as Set 5 (S9 File).
Druggability analysis and identification of novel drug targets
Only 56 proteins showed similarity with the available drug targets, while the remaining 41 showed no hit. These 41 proteins (Set 6) were considered as novel drug targets which include both cytoplasmic and membrane proteins (S10 File). Besides, the results of druggable proteins are provided in S1 Table. Furthermore, the other 41 proteins were considered as novel therapeutic targets and subjected to human ‘anti-targets’ analysis.
‘Anti-target’ analysis and prediction of subcellular localization
A total of 210 ‘anti-targets’ reported in the literature were fetched from NCBI (S11 File). All novel drug target proteins were successfully screened through BLASTp analysis, and no evidence of similarity was seen. Hence, all these novel drug target proteins were listed for human microbiome analysis considering non-homologous to host ‘anti-targets’ (S10 File). Moreover, The results of subcellular localization analysis by four servers are provided in S11 File. The result revealed that among 41 specific proteins involved in pathogen-specific pathways, 16 were cytoplasmic proteins assigned as Set 8 (S12 File, Table 2), while the remaining 25 sequences were membrane proteins.
Table 2. Pathogen specific essential cytoplasmic proteins as novel therapeutic targets.
| Sl. No | Protein Id | KO assignment | Description | Pathways |
|---|---|---|---|---|
| 1 | Q87L83 | K09823 | Fur family transcriptional regulator, zinc uptake regulator | Quorum sensing |
| 2 | Q87GW9 | K07674 | Two-component system, narl family, nitrate/nitrite sensor histidine kinase narq | Two-component system |
| 3 | Q79YW1 | K02410 | Flagellar motor switch protein flig | Bacterial chemotaxis |
| 4 | Q87HD9 | K11904 | Type VI secretion system secreted protein vgrg | Bacterial secretion system |
| 5 | Q87TC9 | K02461 | General secretion pathway protein L | Bacterial secretion system |
| 6 | Q87NV3 | K07780 | Arac family transcriptional regulator required for anaerobic and stationary phase induction of genes | Two-component system |
| 7 | Q87K78 | K07718 | Two-component system, sensor histidine kinase yesm | Two-component system |
| 8 | Q87I65 | K07688 | Two-component system, narl family, response regulator, fimbrial Z protein, fimz | Two-component system |
| 9 | Q87TD5 | K02455 | General secretion pathway protein F | Biofilm formation |
| 10 | Q87MI1 | K03567 | Glycine cleavage system transcriptional repressor | Biofilm formation |
| 11 | Q87HC5 | K11892 | Type VI secretion system protein impk | Bacterial secretion system |
| 12 | Q87Q12 | K02053 | Putative spermidine/putrescine transport system permease protein | Quorum sensing |
| 13 | Q87LE2 | K03092 | RNA polymerase sigma-54 factor | Biofilm formation |
| 14 | Q87HC6 | K11891 | Type VI secretion system protein impl | Biofilm formation |
| 15 | Q79YX4 | K03408 | Purine-binding chemotaxis protein chew | Bacterial chemotaxis |
| 16 | Q87NG0 | K11617 | Two-component system, narl family, sensor histidine kinase lias | Two-component system |
Human microbiome non-homology analysis
Cytoplasmic proteins showing <45% similarity with reported human microbiome proteins were selected for protein-protein interaction analysis, whereas membrane proteins were selected for further vaccine candidacy. However, microbiome analysis revealed that a total of 9 proteins (Set 9) of the pathogen showed <45% similarity with human microflora (S2 Table, S13 File).
Analysis of virulence factors (VF’s) and protein-protein interactions studies (PPIs)
From 9 cytoplasmic novel proteins, five uncharacterized proteins were removed and the remaining four proteins were considered for VFDV analysis. The VFDB result showed that two proteins (Set 10) i.e., VIBPA Type II secretion system protein L (Q87TC9) and VIBPA Putative fimbrial protein Z (Q87I65) were associated with virulence of V. parahaemolyticus (Table 3). These proteins were subjected to protein-protein interaction study. STRING v10.5 revealed that Type II secretion system protein L confers interactions with nine proteins (Fig 3A), while putative fimbrial protein Z exhibits interactions with three other proteins (Fig 3B). These proteins are mainly responsible for protein transport, involved in biofilm formation and bacterial secretion system, or act as regulatory proteins (e.g., transcription regulator, signal transduction response regulator).
Table 3. Predicted therapeutic targets (novel cytoplasmic proteins) showing virulent properties.
Fig 3.

Investigation of PPIs through STRING v10.5 server; (A) UDP-N-acetylmuramoyl-L-alanyl-D-glutamate—2,6-diaminopimelate ligase (murE), (B) Trigger factor (tig).
Screening of drug molecules against novel cytoplasmic proteins
A total of 335 unique metabolites were retrieved from Human Metabolites Database for docking analysis against predicted therapeutic drug targets (novel cytoplasmic proteins). Docking scores were analyzed to screen the top drug candidates with the lowest binding energy (S3 Table). Among top 10 metabolites (Table 4), Eliglustat (DB09039) was found superior in terms of free binding energy for both protein targets, followed by Simvastatin (DB09039) and Hydroxocobalamin (DB00200) for VIBPA Type II secretion system protein L (Q87TC9) and VIBPA Putative fimbrial protein Z (Q87I65) respectively. ADME analysis was performed to get an insight into how the predicted pharmaceuticals will interact with the body as a whole (Table 5).
Table 4. Top 10 metabolites predicted as suitable drug candidates against VIBPA Type II secretion system protein L and VIBPA Putative fimbrial protein Z.
| Novel Targets | HMDB ID | Binding Energy | Name | Drug Bank ID | Drug Name | Drug Group |
|---|---|---|---|---|---|---|
| VIBPA Type II secretion system protein L (Q87TC9) | HMDB0004971 | -10.6 | Glucosylceramide (d18:1/16:0) | DB09039 | Eliglustat | Approved |
| HMDB0004972 | -10.5 | Glucosylceramide (d18:1/18:0) | DB09039 | Eliglustat | Approved | |
| HMDB0004970 | -10.3 | Glucosylceramide (d18:1/9Z-18:1) | DB09039 | Eliglustat | Approved | |
| HMDB0004976 | -10.3 | Glucosylceramide (d18:1/26:1(17Z)) | DB09039 | Eliglustat | Approved | |
| HMDB0008646 | -10.3 | PC(22:4(7Z,10Z,13Z,16Z)/22:4(7Z,10Z,13Z,16Z)) | DB00641 | Simvastatin | Approved | |
| HMDB0011300 | -10.1 | PC(P-18:1(11Z)/P-18:1(11Z) | DB00334 | Olanzipine | Approved, Investigational | |
| HMDB0008443 | -9.7 | PC(20:4(5Z,8Z,11Z,14Z)/20:4(5Z,8Z,11Z,14Z)) | DB00641 | Simvastatin | Approved | |
| HMDB0008138 | -9.6 | PC(18:2(9Z,12Z)/18:2(9Z,12Z)) | DB00641 | Simvastatin | Approved | |
| HMDB0004973 | -9.5 | Glucosylceramide (d18:1/20:0) | DB09039 | Eliglustat | Approved | |
| HMDB0010348 | -9.5 | Dehydroepiandrosterone 3-glucuronide | DB01708 | Prasterone | Approved, Investigationa, Nutraceutical | |
| VIBPA Putative fimbrial protein Z (Q87I65) | HMDB0004972 | -9.6 | Glucosylceramide (d18:1/18:0) | DB09039 | Eliglustat | Approved |
| HMDB0002308 | -9.5 | Hydroxocobalamin | DB00200 | Hydroxocobalamin | Approved | |
| HMDB0004970 | -9.3 | Glucosylceramide (d18:1/9Z-18:1) | DB09039 | Eliglustat | Approved | |
| HMDB0004971 | -9.3 | Glucosylceramide (d18:1/16:0) | DB09039 | Eliglustat | Approved | |
| HMDB0002174 | -9.1 | Cobalamin | DB14098 | Cobalamin | Experimental | |
| HMDB0008646 | -8.9 | PC(22:4(7Z,10Z,13Z,16Z)/22:4(7Z,10Z,13Z,16Z)) | DB00641 | Simvastatin | Approved | |
| HMDB0060546 | -8.9 | Norbuprenorphine | DB01026 | Ketoconazole | Approved, Investigational | |
| HMDB0041936 | -8.7 | Morphine-3-glucuronide | DB00295 | Morphine | Approved, Investigational | |
| HMDB0004974 | -8.6 | Glucosylceramide (d18:1/22:0) | DB09039 | Eliglustat | Approved | |
| HMDB0008443 | -8.5 | PC(20:4(5Z,8Z,11Z,14Z)/20:4(5Z,8Z,11Z,14Z)) | DB00641 | Simvastatin | Approved |
Table 5. ADME profiling of top drug candidates.
| ADME analysis | Top drug candidates | |||
|---|---|---|---|---|
| Eliglustat | Simvastatin | Hydroxocobalamin | ||
| Physicochemical parameters | Formula | C23H36N2O4 | C25H38O5 | C62H89CoN13O15P |
| Molecular weight | 404.54 g/mol | 418.57 g/mol | 1346.36 g/mol | |
| Molar Refractivity | 120.69 | 118.47 | 352.74 | |
| TPSA | 74.52 Å2 | 72.83 Å2 | 468.89 Å2 | |
| Lipophilicity | Log Po/w (iLOGP) | 4.07 | 3.74 | 0.00 |
| Log Po/w (XLOGP3) | 4.02 | 4.68 | -3.48 | |
| Log Po/w (WLOGP) | 3.57 | 4.59 | -0.99 | |
| Log Po/w (MLOGP) | 2.27 | 3.77 | -3.84 | |
| Log Po/w (SILICOS-IT) | 4.88 | 3.77 | -4.08 | |
| Consensus Log Po/w | 3.76 | 4.11 | -2.48 | |
| Log Po/w (iLOGP) | 4.07 | 3.74 | 0.00 | |
| Pharmacokinetics | GI absorption | High | High | Low |
| BBB permeant | Yes | No | No | |
| P-gp substrate | No | No | Yes | |
| CYP1A2 inhibitor | No | No | No | |
| CYP2C19 inhibitor | No | No | No | |
| CYP2C9 inhibitor | No | Yes | No | |
| CYP2D6 inhibitor | Yes | No | No | |
| CYP3A4 inhibitor | No | Yes | No | |
| Log Kp (skin permeation) | -5.91 cm/s | -5.53 cm/s | -16.98 cm/s | |
| Water Solubility | Log S (SILICOS-IT) | -5.05 | -3.56 | -7.11 |
| Solubility | 3.57e-03 mg/ml; 8.83e-06 mol/l | 1.15e-01 mg/ml; 2.74e-04 mol/l | 1.04e-04 mg/ml; 7.73e-08 mol/l | |
| Class | Moderately soluble | Soluble | Poorly soluble | |
| Druglikeness | Bioavailability Score | 0.55 | 0.55 | 0.17 |
| Lipinski | Yes; 0 violation | Yes; 0 violation | No; 3 violations: MW>500, NorO>10, NHorOH>5 | |
| Ghose | Yes | Yes | No; 4 violations: MW>480, WLOGP<-0.4, MR>130, | |
| Veber | No; 1 violation: Rotors>10 | Yes | No; 2 violations: Rotors>10, TPSA>140 | |
| Medicinal Chemistry | Synthetic accessibility http://www.swissadme.ch/index.php | 4.76 | 5.80 | 10.00 |
| PAINS | 0 alert | 0 alert | 0 alert | |
| Brenk | 2 alerts: imine_1, imine_2 | 1 alert: more_than_2_esters | 1 alert: phosphor | |
| Leadlikeness | No; 3 violations: MW>350, Rotors>7, XLOGP3>3.5 | No; 2 violations: MW>350, XLOGP3>3.5 | No; 2 violations: MW>350, Rotors>7 | |
Screening of novel outer membrane proteins for vaccine construction
From the 25 novel outer membrane proteins (S14 File) designated as Set 11, two were selected based on the highest antigenicity score and human microbiome analysis to develop a novel chimeric peptide vaccine against V. parahemolyticus (Table 6). The schematic diagram summarizing the protocol over in silico vaccinomics strategy has been elucidated in Fig 1. Sensor histidine protein kinase (Q87HJ8) and flagellar hook-associated protein (Q87JH9) possessed better antigenicity (0.64 and 0.53 respectively) while showed less percentage similarity (<45% and <41%, respectively) when compared with gut microbiome data (S4 Table).
Table 6. Novel vaccine targets proteins showing higher antigenicity.
T-cell epitope prediction, transmembrane topology screening and antigenicity analysis
A plethora of CTL and HTL epitopes were identified for both proteins that can bind to the different large number of HLA-A and HLA-B alleles using MHC class-I and MHC class II binding predictions of IEDB (S15 File). Top epitopes (MHC-I and MHC-II binding peptides) for both proteins having the capacity to elicit strong T-cell responses were selected as putative T-cell epitope candidates according to their topology screening by TMHMM and antigenic scoring (AS) by Vaxijen server (S16 File).
Population coverage, allergenicity, toxicity and conservancy analysis
Two different population coverages were calculated from CTL and HTL populations for MHC class I and MHC class IIrestricted peptides, respectively (Fig 4). Epitopes, found to be non-allergen for humans, were identified according to the allergenicity assessment via four servers. However, epitopes predicted as a toxin was removed from the proposed list of epitopes. Several epitope candidates from both proteins were found to be highly conserved within different strains of V. parahemolyticus with maximum conservancy level of 99% for histidine protein kinase and 100% for flagellar hook-associated protein respectively (S5 Table). Top 3 epitopes (CTL and HTL) for each protein were considered based on the above mentioned parameters to design the final vaccine construct (Table 7).
Fig 4.
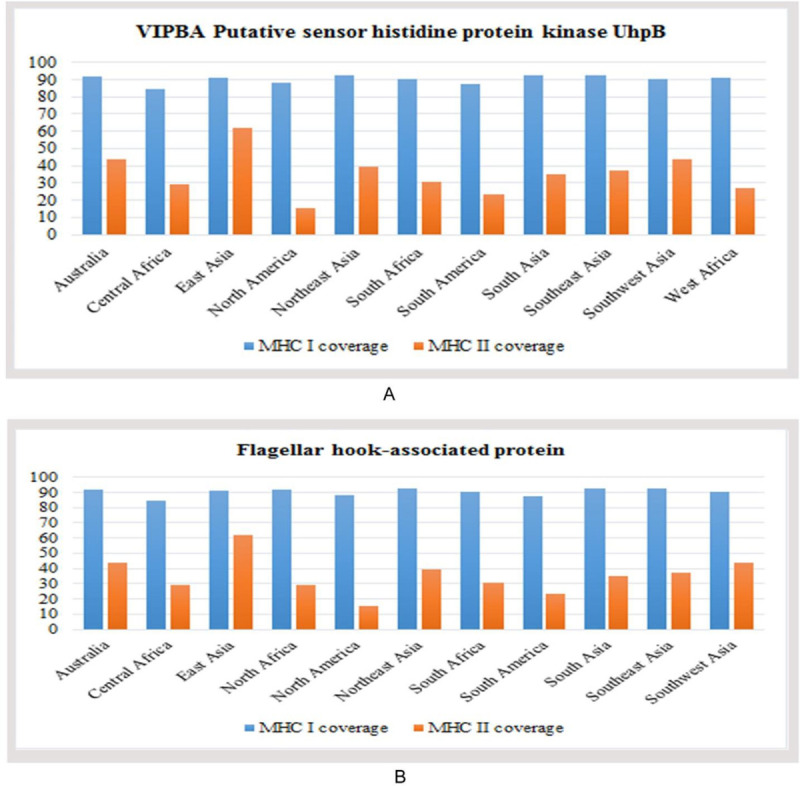
Population coverage analysis of (A) VIPBA putative sensor histidine protein kinase UhpB, and (B) Flagellar hook-associated protein.
Table 7. Predicted final CTL and HTL epitopes of histidine protein kinase and flagellar hook-associated protein.
| Protein | MHC | Epitope | Start | End | Vaxijen | Conservancy |
|---|---|---|---|---|---|---|
| Sensor histidine protein kinase UhpB | MHC-I | AILLFPFAL | 36 | 44 | 2.993 | 99.00% |
| ILLFPFALR | 37 | 45 | 2.9721 | 99.00% | ||
| HDDGVGFKV | 448 | 456 | 2.363 | 99.00% | ||
| MHC-II | ILLFPFALRLGIALH | 37 | 51 | 2.1484 | 87.00% | |
| AILLFPFALRLGIAL | 36 | 50 | 1.9451 | 87.00% | ||
| LLFPFALRLGIALHT | 38 | 52 | 1.8268 | 87.00% | ||
| Flagellar hook-associated protein | MHC-I | FNAQDEEGH | 125 | 133 | 1.8425 | 98.00% |
| GGRHNNLDL | 234 | 242 | 1.7855 | 99.00% | ||
| KPSPNFQAEV | 203 | 212 | 1.4978 | 45.00% | ||
| MHC-II | DSIESSFNAQDEEGH | 199 | 133 | 1.304 | 88.00% | |
| IGGRHNNLDLMDGAH | 233 | 247 | 0.801 | 59.00% | ||
| KLSDDPMASIKLLNL | 38 | 52 | 0.887 | 88.00% |
Prediction of 3D structures for superior epitopes and analysis of molecular docking
The epitopes, showing conservancy pattern at a biologically significant level, were only allowed for further docking analysis. 3D structures were predicted for top epitopes (6 from Flagellar hook-associated protein and six from sensor histidine protein kinase) to analyze their interactions with different HLA alleles. The PEP-FOLD3 server modeled five 3D structures for each epitope, and the best one was identified for docking study. The result showed that ‘AILLFPFAL’ epitope of Flagellar hook-associated protein was superior in terms of free binding energy while interacted with HLA-A* 11:01 (−8.3 kcal/mol). Demonstrated energy was −9.1 kcal/mol for epitope ‘GGRHNNLDL’ of Sensor histidine protein kinase (Table 8).
Table 8. Binding energy of predicted epitopes with selected MHC class I and MHC class II molecules generated from molecular docking by AutoDock.
| Protein | Epitope | MHC Class | Binding Energy |
|---|---|---|---|
| Sensor histidine protein kinase | FNAQDEEGH | HLA-A*11:01 | -8.6 |
| GGRHNNLDL | -9.1 | ||
| KPSPNFQAEV | -8.2 | ||
| DSIESSFNAQDEEGH | HLA-DRB1*04:01 | -7.3 | |
| IGGRHNNLDLMDGAH | -6.5 | ||
| KLSDDPMASIKLLNL | -6.3 | ||
| Flagellar hook-associated protein | AILLFPFAL | HLA-A*11:01 | -8.3 |
| ILLFPFALR | -8.1 | ||
| HDDGVGFKV | -8.1 | ||
| ILLFPFALRLGIALH | HLA-DRB1*04:01 | -7.0 | |
| AILLFPFALRLGIAL | -6.4 | ||
| LLFPFALRLGIALHT | -7.2 |
Identification of B-Cell epitope
B-cell epitopes of both proteins were generated using six different algorithms from IEDB (S1 and S2 Figs). The epitopes were further investigated to reveal their non-allergenicity pattern, and the top one epitope from each prediction was selected for vaccine construction (S6 Table).
Epitope cluster analysis and vaccine construction
The construction of vaccine protein was based on identifying larger cassettes containing multiple epitopes. Epitope cluster analysis tool from IEDB predicted 21 epitope clusters among the top epitopes (6 CTL, 6 HTL epitopes, and 12 BCL epitopes) proposed in Table 8 and S2 Table. Each vaccine construct was occupied by a protein adjuvant, PADRE peptide sequence, T-cell and B-cell epitopes with their respective linkers (S7 Table) Constructs V1, V2 and V3 were 370, 455 and 484 residues long, respectively. PADRE sequence was used to enhance the potency and efficacy of the peptide vaccine.
Allergenicity, antigenicity and solubility prediction of different vaccine constructs
Results revealed V1 as the most potent vaccine candidate with better antigenic nature (1.18) and non-allergic behavior that can elicit a strong immune response (S7 Table). All three constructs showed solubility above the threshold value (0.45). Again, construct V1 was superior in terms of solubility potential. The surface distribution of charge, hydrophobicity and stability were calculated at 91 different combinations of pH and ionic strength (Fig 5).
Fig 5. Solubility prediction of vaccine constructs.
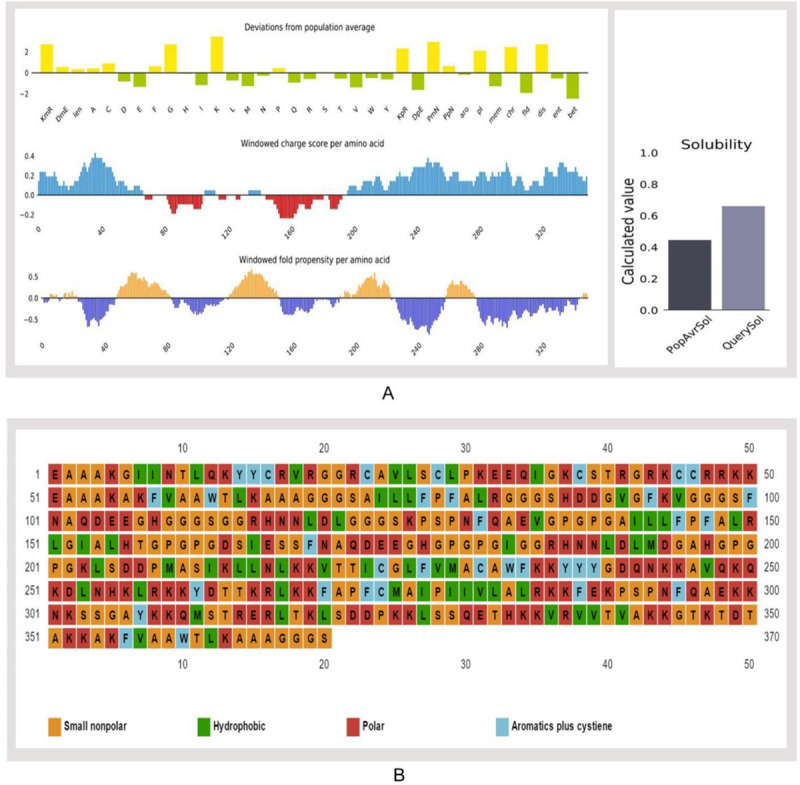
(A) Solubility prediction of designed vaccine construct V1 using via Protein-sol server, and (B) prediction of polar, nonpolar, hydrophobic and aromatic regions.
Physicochemical characterization and secondary structure analysis
The molecular weight of the designed construct V1 was 39.45 kDa. The theoretical pI 9.95 implied that the protein would have a net negative charge above this pI and vice versa. At 0.1% absorption, the extinction coefficient was 26,930, while assuming all cysteine residues reduced. The estimated half-life was predicted to be >10 h in E. coli in vivo, whereas 1 h within mammalian reticulocytes in vitro. Hydrophilic behavior and thermostability of the protein were represented by the GRAVY value and aliphatic index that were −0.510 and 67.62, respectively. Instability index (37.49) and various physicochemical features classified the protein as a stable onewith the capacity to induce a robust immunogenic reaction in the body. The predicted secondary structure confirmed to have 35.6% alpha helix, 11.89% sheet and 52.43% coil region (S3 Fig). Around 34.59% polar, 16.21% hydrophobic and 9.46% aromatic regions were identified in the structure (Fig 5).
Tertiary structure prediction, refinement, validation and disulfide engineering of vaccine construct
I-TASSER predicted five models for each proposed vaccine candidates, which were ranked based on cluster size. Ten best templates (with the highest Z-score) selected from the LOMETS threading programswere used to predict the tertiary structures. Homology modeling was performed by using 1kj6 from RCSB Protein Data Bank) as a best suited template for Vaccine protein V1. Results showed that model 1 had the highest C-Score of -2.11 while the estimated TM-score and RMSD were 0.46±0.15 and 11.6±4.5Å (Fig 6). After refinement, 88.3% and 98.1% residues were in the favored and allowed region revealed by Ramachandran plot analysis (Fig 6). The modeled tertiary structure of designed construct V2 and V3 have been shown in S4 Fig. A total of 22 pairs of amino acid residues were identified with the potential to form disulfide bonds by DbD2 server. However, only two pairs (i.e., ARG 82-Gly 85, Lys 347-Thr 350) were compatible with disulfide bond formation considering the energy, chi3 and B-factor parameter (S5 Fig). All these residues were replaced with a cysteine residue.
Fig 6. Tertiary structure prediction and validation of vaccine protein V1.

(A) Tertiary structure of modeled construct V1, (B) Ramachandran plot analysis of vaccine protein V1.
Protein-protein docking and molecular dynamics simulation
Docking study was conducted between three vaccine constructs (i.e., V1, V2, V3) and different HLA alleles. Construct V1 showed biologically significant results and found to be superior in terms of free binding energy (S8 Table). Besides, the binding affinity of the predicted vaccine and TLR-1/2 heterodimer complex was also analyzed. The 3D structure of human TLR-1/2 heterodimer was retrieved from the RCSB protein data bank. ClusPro generated thirty protein-ligand complexes (clusters) as output along with respective free binding energy. The lowest energy was −1257.9 for cluster 1 (Fig 7). FireDock output refinement of the PatchDock server showed the lowest global energy of −7.08 for solution 5. Normal mode analysis allowed the demonstration of large scale mobility and the stability of proteins. The analysis was performed based on the internal coordinates of the protein-protein complex. In the 3D model, the direction of each residue was given by arrows, and the length of the line represented the extent of mobility (Fig 8A). The eigenvalue found for the complex was 2.4784e−05 (Fig 8B). The vaccine protein V1 and TLR1-2 heterodimers were oriented towards each other. The B-factor values deduced from normal mode analysis was analogous to RMS (Fig 8C). Hinges in the chain indicated the probable deformability of the complex measured by the contortion of each residue (Fig 8D). The variance associated with each normal mode was inversely linked to the eigenvalue. Covariance matrix explained the coupling between pairs of residues was correlated, uncorrelated, or anti-correlated motions were represented via red, white and blue colors, respectively (Fig 8E). The result also generated an elastic network model (Fig 8F) that identified the pairs of atoms connected via springs. Each dot in the diagram was indicated one spring between the corresponding pair of atoms and colored based on the degree of stiffness.
Fig 7. Docked complex of vaccine construct V1 with human TLR 1/2 heterodimer.

(A) Cartoon format, and (B) Ball structure.
Fig 8. Molecular dynamics simulation of vaccine protein V1-TLR8 complex.

Stability of the protein-protein complex was investigated through (A) mobility, (B) B-factor, (C) deformability, (D) eigenvalue, (E) covariance and (F) elastic network analysis.
Codon adaptation, in silico cloning and similarity analysis with human proteins
E. coli strain K12 was selected as the host for the cloning purpose of the vaccine construct V1. Vaccine protein V1 was transcribed reversely, where the Codon Adaptation Index (CAI) was found 0.994, and the GC content of the optimized codons (50.55%) was also significant. The construct did not hold restriction sites for ApaI and BglI, which ensured its safety for cloning purposes. The optimized codons were incorporated into the pET28a(+) vector along with ApaI and BglI restriction sites. A clone of 5634 base pair was obtained, including the 1118 bp desired sequence and the rest belonging to the vector. The desired region was shown in red color in between the pET28a(+) vector sequence (Fig 9). Sequence similarity analysis of the proposed vaccine with human proteins revealed that there was no similarity between predicted vaccine constructs and human proteins.
Fig 9.

In silico restriction cloning of the gene sequence of construct V1 into pET28a(+) expression vector; (A) Restriction digestion of the vector pET28a(+) and construct V1 with BglI and ApaI (B) Inserted desired fragment (V1 Construct) between ApaI (1334) and BglI (2452) indicated in red color.
Discussion
Emergence of rapid antibiotic resistance [35,85], severe effects on human health [14,86], and economic importance for substantially impairing the aquaculture production [4] have made it necessary to identify effective drug targets and vaccine candidates against V. parahaemolyticus. Different computational approaches are now being widely practiced to identify proteins those are essential for the survival of the pathogen and not involved in the metabolic pathways of the host, thereby choosing the proteins associated only in the metabolic pathways of the pathogen is important [38,87]. Essential proteins are most promising for new antibacterial drug targets since most antibiotics are designed to bind essential gene products. Here, subtractive genome approaches (removal of paralogous proteins, identification of non-homologous proteins against the host, identification of essential proteins and metabolic pathways analysis of the pathogen), and vaccinomics strategy were employed for identifying novel drug and vaccine molecules through the comprehensive proteome exploration of V. parahaemolyticus genome.
The complete proteome of V. parahaemolyticus (4822 proteins) was retrieved from the NCBI database, and the homologous proteins were removed based on their identity with human proteins. Proteins encoded by essential genes and unique to an organism can be considered as species-specific drug targets, as they play vital roles in its metabolism. The study revealed 96 unique, essential proteins (Set 5) of V. parahaemolyticus, which can be considered as suitable drug targets for combating V. parahaemolyticus infections. Among the unique proteins, 55 proteins were druggable and can be targeted using existing drugs (92) that are already approved and available in the market (S1 Table). In the case of a broad-spectrum drug, for avoiding mutational changes as well as the emergence of resistant bacteria, the DrugBank databases were screened, which contains entries for 2556 approved small molecule drugs, 962 approved biotech drugs, 112 nutraceuticals and over 5125 experimental drugs. A total of 41 proteins of V. parahaemolyticus showed no similarity after passing through the DrugBank database and listed for the prediction of novel drug targets and vaccine candidates (Set 6). To avoid severe cross-reaction and toxic effects in human, identification of nonhomologous proteins to essential human proteins (referred to as ‘anti-targets’) was a crucial step considered in this study. However, the identified novel drug targets (41) showed no evidence of similarity with the ‘anti-targets’. Although both cytoplasmic and membrane proteins serve the purpose as therapeutic targets [86], membrane proteins are best suited for vaccine candidates [88,89]. Hence, in this study, membrane proteins (25) were used for vaccine construction, whereas cytoplasmic proteins (16) were proposed as suitable drug targets. Targeting human microbiome non-homology proteins is suitable since drugs or vaccines designed and administered for these targets will be less harmful to other commensal microbial strains. Among the novel cytoplasmic proteins (16), only the proteins (9) conferring <45% similarity with human microbiota were retained. The VFDB (virulence factor database) analysis studies confirmed that ‘VIBPA Type II secretion system protein L (Q87TC9)’ and ‘VIBPA Putative fimbrial protein Z (Q87I65)’ were associated with virulence in the host (Set 10). The protein-protein interaction studies also strengthened the superiority of these two proteins as suitable drug targets (Table 4).
Pharmacologically active metabolites can be conveniently used as leads during the lead optimization phase of drug discovery [90–92]. Many drugs are converted to metabolites that can retain the intrinsic affinity of the parent drug for the pharmacological target. The contribution of active drug metabolites efficacy is relative to the contribution of the parent drug, target affinity, functional activity and plasma protein binding [93]. In this study, molecular docking of 350 human metabolites against ‘VIBPA Type II secretion system protein L’ and ‘VIBPA Putative fimbrial protein Z’ was conducted to screen superior drug molecules (S2 Table). The study revealed that ‘Eliglustat (DB09039)’ was the top drug candidate for both protein targets in terms of free binding energy (Table 4). Therefore, it can be suggested as a suitable drug to treat infections caused by V. parahaemolyticus. Eliglustat was first approved in August 2014 by FDA for the treatment of Gaucher's disease [94]. The compound belongs to the class of ‘enzyme inhibitors’ which is believed to work by inhibition of glucosylceramide synthase [95,96]. Eliglustat prevents the formation certain fatty substance in the body which may causes liver, spleen, bone, and blood problems. Mistry et al. reported that oral substrate reduction therapy by Eliglustat resulted in significant improvements in platelet count, liver volume, hemoglobin level and spleen volume, when compared with untreated adults with Gaucher disease [97]. Hydroxocobalamin (vitamin B12), on the contrary, is often used as dietary supplement to treat pernicious anemia, cyanide poisoning, toxic amblyopia and Leber's optic atrophy [98,99]. However, antibacterial activity of vitamin B12 is not new. There is also experimental evidence of synergistic antimicrobial effects of vitamin B12 with other antibiotics [100]. In the present study Hydroxocobalamin showed significantly higher binding affinity to VIBPA Putative fimbrial protein Z (Table 4). Therefore, it can be used as supplement with other drugs to combat V. Parahemolyticus associated infections. Simvastatin (belongs to the group of medicines called statins) is another approved drug which is often used to treat patients with high blood cholesterol. It is also prescribed against rheumatoid arthritis, type-1 or type-2 diabetes [101,102], and to prevent strokes and heart attacks [103,104].
Recently, the possibility of Simvastatin as a therapeutic option for COVID-19 was reported due to its ability to block the key factors required for infectivity [105,106]. Usually, it is considered to be a very safe medicine and unusual to have side effects [107]. However, cautions should be taken for the repurposed use of these drugs to minimize the risk of potential toxicity.
Moreover, drug profiling (Physicochemical parameters, Lipophilicity, Pharmacokinetics, Water Solubility, Druglikeness, Medicinal Chemistry) of ‘Eliglustat (DB09039)’ along with other top candidates, i.e., ‘Simvastatin (DB09039)’ and ‘Hydroxocobalamin (DB00200)’ were also performed through ADME analysis (Table 5), which showed no undesirable effects that could reduce their drug-likeness properties.
Several advantages help the researchers to select membrane proteins both as drug and vaccine candidates as their functions can be easily studied through computer-based approaches than wet-lab process [89,108]. In this study, two vaccine targets, ‘Sensor histidine protein kinase UhpB (Q87HJ8)’ and ‘Flagellar hook-associated protein (Q87JH9)’ were selected after screening the novel outer-membrane proteins (25) based on their antigenicity score and human microbiome non-homology analysis (Table 6). Both the proteins further analyzed to design a potent, highly immunogenic vaccine candidate against V. parahaemolyticus. Numerous antigenic epitopes were generated which were investigated extensively for antigenicity, allergenicity, toxicity, conservancy and other physiochemical properties using a number of bioinformatics tools and software. The final vaccine constructs were designed with the help of different adjuvants and amino acid linkers [84]. It has been reported that the PADRE sequence reduces the polymorphism of HLA molecules in the population [108,109]. Linkers in vaccines also enhanced the immunogenicity of the vaccines in previous studies [110,111]. Therefore, all the important that could induce the immunogenicity of the designed vaccine constructs were taken. Also, disulfide engineering was employed to enhance the stability of the designed vaccine. The purpose of the molecular docking analysis was to show the proposed epitopes could interact with at least one MHC molecule at minimum binding energy. Therefore, it was done to explore the binding affinity of promiscuous epitopes with different HLA alleles including HLA-DRB1*03:01 (1A6A), (HLA-DRB5*01:01 (1H15), HLA-DRB3*01:01 (2Q6W), HLA-DRB1*04:01 (2SEB), HLA-DRB1*01:01 (2FSE), and HLA-DRB3*02:02 (3C5J). The OmpU, one of the major outer membrane porins of V. parahaemolyticus, is recognized by the Toll-like receptor 1/2 (TLR-1/2) heterodimer in THP-1 monocytes [112]. So, a docking study was also performed to analyze the affinity between the designed construct and human TLR-1/2 heterodimer. The vaccine receptor complex showed deformability at a minimum level, which also strengthened our prediction. Finally, the optimized codons of the designed construct been cloned into the pET28a(+) vector of E. coli strain K12.
The idea of subtractive genomic analysis using various bioinformatics tools has brought a revolution in the drug discovery process. The present study will help to develop novel therapeutics and preventive measures against V. parahaemolyticus, thereby help to reduce the mortality and morbidity caused by it. However, further in vivo trials using model organisms are highly recommended to validate our prediction.
Supporting information
B-cell epitope prediction of Sensor histidine protein kinase UhpB (A: Linear, B: Beta-turn, C: Flexibility, D: Surface Accessibility, E: Antigenicity, F: Hydrophilicity). For each graph, X-axis and Y-axis represent the position and score. Residues that fall above the threshold value are shown in yellow color while the highest peak in yellow color identifies most favored position.
(PPTX)
B-cell epitope prediction of Flagellar hook-associated protein (A: Linear, B: Beta-turn, C: Flexibility, D: Surface Accessibility, E: Antigenicity, F: Hydrophilicity). For each graph, X-axis and Y-axis represent the position and score. Residues that fall above the threshold value are shown in yellow color while the highest peak in yellow color identifies most favored position.
(PPTX)
(PPTX)
(A) V2 and (B) V3 generated via RaptorX server.
(PPTX)
(A) Initial form, (B) Mutant form.
(PPTX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOC)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Kuwaiti A Al, Subbarayalu AV. Vibrio parahaemolyticus A review on distribution, pathogenesis, virulence determinants and epidemiology. Saudi J Med Med Sci. 2017; 260–267. 10.4103/sjmms.sjmms_98_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen AJ, Hasan NA, Haley BJ, Taviani E, Tarnowski M, Brohawn K, et al. Characterization of pathogenic Vibrio parahaemolyticus from the Chesapeake Bay, Maryland. Front Microbiol. 2017;8: 1–10. 10.3389/fmicb.2017.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Letchumanan V, Chan KG, Lee LH. Vibrio parahaemolyticus: A review on the pathogenesis, prevalence, and advance molecular identification techniques. Front Microbiol. 2014;5: 1–13. 10.3389/fmicb.2014.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rico A, Satapornvanit K, Haque MM, Min J, Nguyen PT, Telfer TC, et al. Use of chemicals and biological products in Asian aquaculture and their potential environmental risks: A critical review. Rev Aquac. 2012;4: 75–93. 10.1111/j.1753-5131.2012.01062.x [DOI] [Google Scholar]
- 5.Bondad-Reantaso MG, Subasinghe RP, Arthur JR, Ogawa K, Chinabut S, Adlard R, et al. Disease and health management in Asian aquaculture. Food Agric Organ. 2016;132: 249–272. 10.1016/j.vetpar.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 6.Wall BA, Mateus A, Marshall L, Pfeiffer D, Lubroth J, Ormel HJ, et al. Drivers, Dynamics and Epidemiology of Antimicrobial Resistance in Animal Production. Food and Agriculture Organization. 2016. [Google Scholar]
- 7.Tendencia EA, De La Peña LD. Antibiotic resistance of bacteria from shrimp ponds. Aquaculture. 2001;195: 193–204. 10.1016/S0044-8486(00)00570-6 [DOI] [Google Scholar]
- 8.Yang Y, Xie J, Li H, Tan S, Chen Y, Yu H. Prevalence, antibiotic susceptibility and diversity of Vibrio parahaemolyticus isolates in seafood from South China. Front Microbiol. 2017;8: 1–9. 10.3389/fmicb.2017.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osunla CA, Okoh AI. Vibrio pathogens: A public health concern in rural water resources in sub-Saharan Africa. Int J Environ Res Public Health. 2017;14: 1–27. 10.3390/ijerph14101188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung SW. A foodborne outbreak of gastroenteritis caused by Vibrio parahaemolyticus associated with cross-contamination from squid in Korea. Epidemiol Health. 2018;40: e2018056 10.4178/epih.e2018056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Tam YH, Yuan J, Chen F, Cai W, Liu J, et al. A foodborne outbreak of gastroenteritis caused by Vibrio parahaemolyticus and norovirus through non-seafood vehicle. PLoS One. 2015;10: 4–11. 10.1371/journal.pone.0137848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniels NA, MacKinnon L, Bishop R, Altekruse S, Ray B, Hammond RM, et al. Vibrio parahaemolyticus Infections in the United States, 1973–1998. J Infect Dis. 2000;181: 1661–1666. 10.1086/315459 [DOI] [PubMed] [Google Scholar]
- 13.Jahangir Alam M, Tomochika KI, Miyoshi SI, Shinoda S. Environmental investigation of potentially pathogenic Vibrio parahaemolyticus in the Seto-Inland Sea, Japan. FEMS Microbiol Lett. 2002;208: 83–87. 10.1111/j.1574-6968.2002.tb11064.x [DOI] [PubMed] [Google Scholar]
- 14.Wu Y, Wen J, Ma Y, Ma X, Chen Y. Epidemiology of foodborne disease outbreaks caused by Vibrio parahaemolyticus, China, 2003–2008. Food Control. 2014;46: 197–202. 10.1016/j.foodcont.2014.05.023 [DOI] [Google Scholar]
- 15.Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin Microbiol Rev. 2007;20: 39–48. 10.1128/CMR.00025-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chao G, Wang F, Zhou X, Jiao X, Huang J, Pan Z, et al. Origin of Vibrio parahaemolyticus O3:K6 pandemic clone. Int J Food Microbiol. 2011;145: 459–463. 10.1016/j.ijfoodmicro.2011.01.022 [DOI] [PubMed] [Google Scholar]
- 17.Newton A, Kendall M, Vugia DJ, Henao OL, Barbara E, Branch DE, et al. Increasing rates of Vibriosis in the UnitedStates, 1996–2010: review of surveillance data from 2 systems. Clin Infect Dis. 2015;54 10.1093/cid/cis243.Increasing [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang JY, Henao OL, Griffin PM, Vugia DJ, Cronquist AB, Hurd S, et al. Infection with pathogens transmitted commonly through food and the effect of increasing use of culture-independent diagnostic tests on surveillance—Foodborne diseases active surveillance network, 10 U.S. sites, 2012–2015. Morb Mortal Wkly Rep. 2016;65: 368–371. 10.15585/mmwr.mm6514a2 [DOI] [PubMed] [Google Scholar]
- 19.Cruz CD, Hedderley D, Fletcher GC. Long-term study of Vibrio parahaemolyticus prevalence and distribution in New Zealand shellfish. Appl Environ Microbiol. 2015;81: 2320–2327. 10.1128/AEM.04020-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Letchumanan V, Yin WF, Lee LH, Chan KG. Prevalence and antimicrobial susceptibility of Vibrio parahaemolyticus isolated from retail shrimps in Malaysia. Front Microbiol. 2015;6: 1–11. 10.3389/fmicb.2015.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gode-potratz CJ, Kustusch RJ, Breheny PJ, Weiss DS, Mccarter LL. Surface sensing in Vibrio parahaemolyticus triggers a program of gene expression that promotes colonization and virulence.Mol Microbiol. Gode-Potratz.2011.pdf. 2012;79: 240–263. 10.1111/j.1365-2958.2010.07445.x.Surface [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, Zhong Y, Gu X, Yuan J, Saeed AF, Wang S. The pathogenesis, detection, and prevention of Vibrio parahaemolyticus. Front Microbiol. 2015;6: 1–13. 10.3389/fmicb.2015.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahimi E, Ameri M, Doosti A, Gholampour AR. Occurrence of toxigenic Vibrio parahaemolyticus strains in shrimp in Iran. Foodborne Pathog Dis. 2010;7: 1107–1111. 10.1089/fpd.2010.0554 [DOI] [PubMed] [Google Scholar]
- 24.Ham H, Orth K. The role of type III secretion System 2 in Vibrio parahaemolyticus pathogenicity. J Microbiol. 2012;50: 719–725. 10.1007/s12275-012-2550-2 [DOI] [PubMed] [Google Scholar]
- 25.Ahmad A, Brumble L, Maniaci M. Vibrio parahaemolyticus Induced Necrotizing Fasciitis: An Atypical Organism Causing an Unusual Presentation. Case Rep Infect Dis. 2013;2013: 1–4. 10.1155/2013/216854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Orth K. Virulence determinants for Vibrio parahaemolyticus infection. Curr Opin Microbiol. 2013;16: 70–77. 10.1016/j.mib.2013.02.002 [DOI] [PubMed] [Google Scholar]
- 27.Xiao J, Liu L, Ke Y, Li X, Liu Y, Pan Y, et al. Shrimp AHPND-causing plasmids encoding the PirAB toxins as mediated by pirAB-Tn903 are prevalent in various Vibrio species. Sci Rep. 2017;7: 1–11. 10.1038/s41598-016-0028-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu WT, Jong KJ, Lin YR, Tsai S en, Tey YH, Wong H chung. Prevalence of Vibrio parahaemolyticus in oyster and clam culturing environments in Taiwan. Int J Food Microbiol. 2013;160: 185–192. 10.1016/j.ijfoodmicro.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 29.Liu H. Analysis of the collective food poisoning events in Shanghai from 1990 to 2000. Chinese J Nat Med. 2003;5: 17–20. 10.16309/j.cnki.issn.1007-1776.2003.03.004 [DOI] [Google Scholar]
- 30.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, et al. Genome sequence of Vibrio parahaemolyticus: A pathogenic mechanism distinct from that of V cholerae. Lancet. 2003;361: 743–749. 10.1016/S0140-6736(03)12659-1 [DOI] [PubMed] [Google Scholar]
- 31.Hiyoshi H, Kodama T, Iida T, Honda T. Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect Immun. 2010;78: 1772–1780. 10.1128/IAI.01051-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jun JW, Shin TH, Kim JH, Shin SP, Han JE, Heo GJ, et al. Bacteriophage therapy of a Vibrio parahaemolyticus infection caused by a multiple-antibiotic-resistant O3:K6 pandemic clinical strain. J Infect Dis. 2014;210: 72–78. 10.1093/infdis/jiu059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin DM, Koskella B, Lin HC. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J Gastrointest Pharmacol Ther. 2017;8: 162 10.4292/wjgpt.v8.i3.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sudha S, Mridula C, Silvester R, Hatha AAM. Prevalence and antibiotic resistance of pathogenic Vibrios in shellfishes from Cochin market. Indian J Geo-Marine Sci. 2014;43: 815–824. [Google Scholar]
- 35.Lee LH, Mutalib NSA, Law JWF, Wong SH, Letchumanan V. Discovery on antibiotic resistance patterns of Vibrio parahaemolyticus in Selangor reveals carbapenemase producing Vibrio parahaemolyticus in marine and freshwater fish. Front Microbiol. 2018;9: 1–13. 10.3389/fmicb.2018.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barh D, Tiwari S, Jain N, Ali A, Santos AR, Misra AN, et al. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev Res. 2011;72: 162–177. 10.1002/ddr.20413 [DOI] [Google Scholar]
- 37.Azim KF, Hasan M, Hossain MN, Somana SR, Hoque SF, Bappy MNI, et al. Immunoinformatics approaches for designing a novel multi epitope peptide vaccine against human norovirus (Norwalk virus). Infect Genet Evol. 2019;74: 103936 10.1016/j.meegid.2019.103936 [DOI] [PubMed] [Google Scholar]
- 38.Hosen MI, Tanmoy AM, Mahbuba D Al, Salma U, Nazim M, Islam MT, et al. Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11. Interdiscip Sci Comput Life Sci. 2014;6: 48–56. 10.1007/s12539-014-0188-y [DOI] [PubMed] [Google Scholar]
- 39.Barh D, Kumar A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol. 2009;9: 225–231. 10.3233/ISB-2009-0399 [DOI] [PubMed] [Google Scholar]
- 40.Owens J. Determining druggability. Nat Rev Drug Discov. 2007;6: 187–187. 10.1038/nrd2275 [DOI] [Google Scholar]
- 41.Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophys Rev. 2017;9: 91–102. 10.1007/s12551-016-0247-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015. 10.3390/molecules200713384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Godzik A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22: 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 44.Luo H, Lin Y, Gao F, Zhang CT, Zhang R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42: 574–580. 10.1093/nar/gkt1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35: 182–185. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46: D1074–D1082. 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The Human Microbiome Project. Nature. 2007;449: 804–810. 10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33: 325–328. 10.1093/nar/gki177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47: D607–D613. 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anil KTJW. Autodock vina: improving the speed and accuracy of docking. J Comput Chem. 2019;31: 455–461. 10.1002/jcc.21334.AutoDock [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doytchinova IA, Flower DR. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8: 1–7. 10.1186/1471-2105-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305: 567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 53.Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43: D405–D412. 10.1093/nar/gku938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30: 846–851. 10.1093/bioinformatics/btt619 [DOI] [PubMed] [Google Scholar]
- 55.Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v.2 - A server for in silico prediction of allergens. J Mol Model. 2014;20 10.1007/s00894-014-2278-5 [DOI] [PubMed] [Google Scholar]
- 56.Fiers MWEJ, Kleter GA, Nijland H, Peijnenburg AACM, Nap JP, van Ham RCHJ. AllermatchTM, a webtool for the prediction of potential allergenicity according to current FAO/WHO Codex alimentarius guidelines. BMC Bioinformatics. 2004;5: 1–6. 10.1186/1471-2105-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodman RE, Ebisawa M, Ferreira F, Sampson HA, van Ree R, Vieths S, et al. AllergenOnline: A peer-reviewed, curated allergen database to assess novel food proteins for potential cross-reactivity. Mol Nutr Food Res. 2016;60: 1183–1198. 10.1002/mnfr.201500769 [DOI] [PubMed] [Google Scholar]
- 58.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GPS. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS One. 2013;8 10.1371/journal.pone.0073957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Li. Application of an improved loop-mediated isothermal amplification detection of Vibrio parahaemolyticus from various seafood samples. African J Microbiol Res. 2011;5 10.5897/ajmr11.1237 [DOI] [Google Scholar]
- 60.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J …. 2010;30: 2785–2791. 10.1002/jcc.21256.AutoDock4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276: 172–174. 10.1016/0014-5793(90)80535-q [DOI] [PubMed] [Google Scholar]
- 62.Karplus PA, Schulz GE. Prediction of chain flexibility in proteins—A tool for the selection of peptide antigens. Naturwissenschaften. 1985;72: 212–213. 10.1007/BF01195768 [DOI] [Google Scholar]
- 63.Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45: W24–W29. 10.1093/nar/gkx346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emini EA, Hughes J V, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55: 836–839. 10.1128/JVI.55.3.836-839.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parker JMR, Guo D, Hodges RS. New Hydrophilicity Scale Derived from High-Performance Liquid Chromatography Peptide Retention Data: Correlation of Predicted Surface Residues with Antigenicity and X-ray-Derived Accessible Sites. Biochemistry. 1986;25: 5425–5432. 10.1021/bi00367a013 [DOI] [PubMed] [Google Scholar]
- 66.Chou PY, Fasman GD. Prediction of the Secondary Structure of Proteins From Their Amino Acid Sequence. Adv Enzymol Relat Areas Mol Biol. 2006;47: 45–148. 10.1002/9780470122921.ch2 [DOI] [PubMed] [Google Scholar]
- 67.Rana A, Akhter Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology. 2016;221: 544–557. 10.1016/j.imbio.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 68.Saha S, Raghava GPS. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34: 202–209. 10.1093/nar/gkl343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein-Sol: A web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33: 3098–3100. 10.1093/bioinformatics/btx345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hasan M, Joy ZF, Bhuiyan EH, Islam MS. In silico characterization and motif election of neurotoxins from snake venom. Am J Biochem Biotechnol. 2015;11: 84–91. 10.3844/ajbbsp.2015.84.91 [DOI] [Google Scholar]
- 71.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112: 531–552. 10.1385/1-59259-584-7:531 [DOI] [PubMed] [Google Scholar]
- 72.Kosciolek T, Jones DT. De novo structure prediction of globular proteins aided by sequence variation-derived contacts. PLoS One. 2014;9 10.1371/journal.pone.0092197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101: 2525–2534. 10.1016/j.bpj.2011.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 2010;66: 12–21. 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Craig DB, Dombkowski AA. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14: 0–6. 10.1186/1471-2105-14-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32: 96–99. 10.1093/nar/gkh354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Macalino SJY, Basith S, Clavio NAB, Chang H, Kang S, Choi S. Evolution of in silico strategies for protein-protein interaction drug discovery. Molecules. 2018;23 10.3390/molecules23081963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33: 363–367. 10.1093/nar/gki481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008;36: 229–232. 10.1093/nar/gkn186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P. IMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42: 271–276. 10.1093/nar/gku339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cui Q, Bahar I. Normal mode analysis theoretical and applications to biological and chemical systems. Brief Bioinform. 2007;8 10.1093/bib/bbm010 [DOI] [Google Scholar]
- 82.Prabhakar PK, Srivastava A, Rao KK, Balaji P V. Monomerization alters the dynamics of the lid region in Campylobacter jejuni CstII: An MD simulation study. J Biomol Struct Dyn. 2016;34: 778–791. 10.1080/07391102.2015.1054430 [DOI] [PubMed] [Google Scholar]
- 83.Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33: 526–531. 10.1093/nar/gki376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Solanki V, Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci Rep. 2018;8: 1–19. 10.1038/s41598-017-17765-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Velazquez-Roman J, León-Sicairos N, Hernández-Díaz L de J, Canizalez-Roman A. Pandemic Vibrio parahaemolyticus O3: K6 on the American continent. Front Cell Infect Microbiol. 2014;3: 1–14. 10.3389/fcimb.2013.00110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Michael CA, Dominey-Howes D, Labbate M. The antimicrobial resistance crisis: Causes, consequences, and management. Front Public Heal. 2014;2: 1–8. 10.3389/fpubh.2014.00145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hossain M, Chowdhury DUS, Farhana J, Akbar MT, Chakraborty A, Islam S, et al. Identification of potential targets in Staphylococcus aureus N315 using computer aided protein data analysis. Bioinformation. 2013;9: 187–192. 10.6026/97320630009187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baliga P, Shekar M, Venugopal MN. Potential Outer Membrane Protein Candidates for Vaccine Development Against the Pathogen Vibrio anguillarum: A Reverse Vaccinology Based Identification. Curr Microbiol. 2018;75: 368–377. 10.1007/s00284-017-1390-z [DOI] [PubMed] [Google Scholar]
- 89.Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J, et al. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog. 2019;130: 19–37. 10.1016/j.micpath.2019.02.023 [DOI] [PubMed] [Google Scholar]
- 90.Clader JW. The Discovery of Ezetimibe: A View from Outside the Receptor. J Med Chem. 2004;47: 1–9. 10.1021/jm030283g [DOI] [PubMed] [Google Scholar]
- 91.Rosenblum SB, Huynh T, Afonso A, Davis HR, Yumibe N, Clader JW, et al. A Designed, Potent, Orally Active Inhibitor of Cholesterol Absorption. J Med Chem. 1998;41: 973–980. 10.1021/jm970701f [DOI] [PubMed] [Google Scholar]
- 92.Van Heek M, France CF, Compton DS, et al. In vivo metabolism-based discovery of a potent cholesterol absorption inhibitor, SCH58235, in the rat and rhesus monkey through the identification of the active metabolites of SCH48461. J Pharmacol Exp Ther. 1997;283: 157–163. [PubMed] [Google Scholar]
- 93.Scott Obach R. Pharmacologically active drug metabolites: Impact on drug discovery and pharmacotherapy. Pharmacol Rev. 2013;65: 578–640. 10.1124/pr.111.005439 [DOI] [PubMed] [Google Scholar]
- 94.Bennett LL, Turcotte K. Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease. Drug Des Devel Ther. 2015;9: 4639–4647. 10.2147/DDDT.S77760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abe A, Inokuchi J ichi, Jimbo M, Shimeno H, Nagamatsu A, Shayman JA, et al. Improved inhibitors of glucosylceramide synthase. J Biochem. 1992;111: 191–196. 10.1093/oxfordjournals.jbchem.a123736 [DOI] [PubMed] [Google Scholar]
- 96.Shayman JA. Eliglustat Tartrate: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease. Drugs Futur 2010. 2010;35: 613–620. 10.1038/jid.2014.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mistry PK, Lukina E, Ben Turkia H, et al. Effect of Oral Eliglustat vs Placebo on Spleen Volume in Patients with Splenomegaly and Gaucher Disease Type 1: The ENGAGE Randomized Clinical Trial. JAMA. 2015;313: 695–706. 10.1001/jama.2015.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maclennan L, Moiemen N. Management of cyanide toxicity in patients with burns. Burns. 2015;41: 18–24. 10.1016/j.burns.2014.06.001 [DOI] [PubMed] [Google Scholar]
- 99.Marc C. Stuart, Maria Kouimtzi SRH. World Health Organization: WHO Model Formulary 2008 Marc C. Stuart, Maria Kouimtzi SRH, editor. Pharmaceutical Journal. WHO; 2009. [Google Scholar]
- 100.Shahzad S, Ashraf MA, Sajid M, Shahzad A, Rafique A, Mahmood MS. Evaluation of synergistic antimicrobial effect of vitamins (A, B1, B2, B6, B12, C, D, E and K) with antibiotics against resistant bacterial strains. J Glob Antimicrob Resist. 2018;13: 231–236. 10.1016/j.jgar.2018.01.005 [DOI] [PubMed] [Google Scholar]
- 101.Hermann F, Forster A, Chenevard R, Enseleit F, Hürlimann D, Corti R, et al. Simvastatin improves endothelial function in patients with rheumatoid arthritis. J Am Coll Cardiol. 2005;45: 461–464. 10.1016/j.jacc.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 102.Sen K, Misra A, Kumar A, Pandey RM. Simvastatin retards progression of retinopathy in diabetic patients with hypercholesterolemia. Diabetes Res Clin Pract. 2002;56: 1–11. 10.1016/s0168-8227(01)00341-2 [DOI] [PubMed] [Google Scholar]
- 103.Minder CM, Blaha MJ, Horne A, Michos ED, Kaul S, Blumenthal RS. Evidence-based use of statins for primary prevention of cardiovascular disease. Am J Med. 2012;125: 440–446. 10.1016/j.amjmed.2011.11.013 [DOI] [PubMed] [Google Scholar]
- 104.Young-Xu Y, Jabbour S, Goldberg R, Blatt CM, Graboys T, Bilchik B, et al. Usefulness of statin drugs in protecting against atrial fibrillation in patients with coronary artery disease. Am J Cardiol. 2003;92: 1379–1383. 10.1016/j.amjcard.2003.08.040 [DOI] [PubMed] [Google Scholar]
- 105.Azim KF, Ahmed SR, Banik A, Khan MMR, Deb A, Somana SR. Screening and druggability analysis of some plant metabolites against SARS-CoV-2: An integrative computational approach. Informatics Med Unlocked. 2020;20: 100367 10.1016/j.imu.2020.100367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jung K, Wang Q, Kim Y, Scheuer K, Zhang Z, Shen Q, et al. The effects of simvastatin or interferon-α on infectivity of human norovirus using a gnotobiotic pig model for the study of antivirals. PLoS One. 2012;7 10.1371/journal.pone.0041619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scott RS, Lintott CJ WM. Simvastatin and Stroke. N Z Med J. 1991;104: 493–495. [PubMed] [Google Scholar]
- 108.Hasan M, Islam S, Chakraborty S, Mustafa AH, Azim KF, Joy ZF, et al. Contriving a chimeric polyvalent vaccine to prevent infections caused by herpes simplex virus (type-1 and type-2): an exploratory immunoinformatic approach. J Biomol Struct Dyn. 2019;0: 000. 10.1080/07391102.2019.1647286 [DOI] [PubMed] [Google Scholar]
- 109.Ghaffari-Nazari H, Tavakkol-Afshari J, Jaafari MR, Tahaghoghi-Hajghorbani S, Masoumi E, Jalali SA. Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T Help in BALB/c mice. PLoS One. 2015;10: 1–12. 10.1371/journal.pone.0142563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hasan M, Azim KF, Begum A, Khan NA, Shammi TS, Imran AS, et al. Vaccinomics strategy for developing a unique multi-epitope monovalent vaccine against Marburg marburgvirus. Infect Genet Evol. 2019;70: 140–157. 10.1016/j.meegid.2019.03.003 [DOI] [PubMed] [Google Scholar]
- 111.Yang Y, Sun W, Guo J, Zhao G, Sun S, Yu H, et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum Vaccines Immunother. 2015;11: 795–805. 10.1080/21645515.2015.1012017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gulati A, Kumar R, Mukhopadhaya A. Differential recognition of Vibrio parahaemolyticus OmpU by Toll-like receptors in monocytes and macrophages for the induction of proinflammatory responses. Infect Immun. 2019;87: 1–22. 10.1128/IAI.00809-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
B-cell epitope prediction of Sensor histidine protein kinase UhpB (A: Linear, B: Beta-turn, C: Flexibility, D: Surface Accessibility, E: Antigenicity, F: Hydrophilicity). For each graph, X-axis and Y-axis represent the position and score. Residues that fall above the threshold value are shown in yellow color while the highest peak in yellow color identifies most favored position.
(PPTX)
B-cell epitope prediction of Flagellar hook-associated protein (A: Linear, B: Beta-turn, C: Flexibility, D: Surface Accessibility, E: Antigenicity, F: Hydrophilicity). For each graph, X-axis and Y-axis represent the position and score. Residues that fall above the threshold value are shown in yellow color while the highest peak in yellow color identifies most favored position.
(PPTX)
(PPTX)
(A) V2 and (B) V3 generated via RaptorX server.
(PPTX)
(A) Initial form, (B) Mutant form.
(PPTX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOC)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.