

Abstract
Cellular infections by DNA viruses trigger innate immune responses mediated by DNA sensors. The cyclic GMP–AMP synthase (cGAS)‐stimulator of interferon gene (STING) signaling pathway has been identified as a DNA‐sensing pathway that activates interferons in response to viral infection and, thus, mediates host defense against viruses. Previous studies have identified oncogenes E7 and E1A of the DNA tumor viruses, human papillomavirus 18 (HPV18) and adenovirus, respectively, as inhibitors of the cGAS‐STING pathway. However, the function of STING in infected cells and the mechanism by which HPV18 E7 antagonizes STING‐induced Interferon beta production remain unknown. We report that HPV18 E7 selectively antagonizes STING‐triggered nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) activation but not IRF3 activation. HPV18 E7 binds to STING in a region critical for NF‐κB activation and blocks the nuclear accumulation of p65. Moreover, E7 inhibition of STING‐triggered NF‐κB activation is related to HPV pathogenicity but not E7–Rb binding. HPV18 E7, severe acute respiratory syndrome coronavirus‐2 open reading frame 3a, human immunodeficiency virus‐2 viral protein X, and Kaposi's sarcoma‐associated herpesvirus KSHV viral interferon regulatory factor 1 selectively inhibited STING‐triggered NF‐κB or IRF3 activation, suggesting a convergent evolution among these viruses toward antagonizing host innate immunity. Collectively, selective suppression of the cGAS‐STING pathway by viral proteins is likely to be a key pathogenic determinant, making it a promising target for treating oncogenic virus‐induced tumor diseases.
Keywords: cGAS‐STING pathway, convergent evolution, DNA tumor virus, HPV18 E7
1. INTRODUCTION
Various nucleic acid sensors converge on the STING adapter protein, which induces type I IFN expression via IRF3 and nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) nuclear translocation, and IFN‐stimulated gene expression. 1 , 2 , 3 Several DNA and RNA viruses antagonize the GMP–AMP synthase (cGAS)‐stimulator of interferon gene (STING) pathway. 4 , 5 Notably, different viral proteins have different mechanisms for antagonizing STING, 6 and the variety of immune evasion strategies employed by different viruses indicates the importance of this pathway in sensing viral pathogens. 6
Human papillomaviruses (HPVs) are small double‐stranded DNA viruses that cause over 99% of cervical cancers. 7 , 8 The growth of HPV‐transformed cells, especially those with high‐risk HPV16/18, depends on the ability of the viral oncoproteins E6 and E7 to manipulate genomic instability and the signaling pathways involved in cell proliferation and cell death. 9 , 10 E6 and E7 inactivate the p53 and Rb tumor suppressor pathways, respectively, 10 , 11 and both high‐ and low‐risk HPV E7 proteins can interact with Rb via Leu‐X‐Cys‐X‐Glu (LXCXE) motifs. 11 , 12 , 13 Why DNA tumor viruses have evolved oncogenes is a long‐standing question. 14
Lau et al. first identified DNA tumor viral oncogenes, including HPV18 E7 and adenovirus E1A, as potent and specific inhibitors of the cGAS‐STING pathway. 15 E1A and E7 bind to STING, and silencing of these oncogenes in human tumor cells restores the cGAS‐STING pathway, revealing a host–virus conflict that may have shaped the evolution of viral oncogenes. 15 , 16 Kaposi's sarcoma‐associated herpesvirus (KSHV) is a DNA virus linked to human malignancies, 17 , 18 and KSHV viral interferon regulatory factor 1 (vIRF1) inhibits cGAS‐STING‐dependent Interferon beta (IFNβ) induction. 17 , 18 However, there is still much to be uncovered regarding the function of STING within infected cells. A better understanding of the interactions between oncovirus proteins and the human host should help in the prevention of tumor diseases induced by oncogenic viruses and inform the development of treatments. 6
Herein, we demonstrated that HPV18 E7 selectively antagonizes cGAS‐STING‐triggered NF‐κB activation but not IRF3 activation. HPV18 E7 binds to STING in a unique region critical for NF‐κB activation and blocks the nuclear accumulation of p65, thereby inhibiting NF‐κB signaling. Additionally, we found that high‐risk tumor‐inducing E7 oncoproteins (HPV18 E7), but not low‐risk tumor‐inducing E7 oncoproteins (HPV6 E7 and HPV11 E7), bound to STING and inhibited STING‐triggered NF‐κB activation. E7 antagonizes STING and is related to HPV pathogenicity. Interestingly, we found that the HPV18 E7 mutant, in which Rb binding was disrupted, still had the ability to antagonize STING‐triggered NF‐κB activation. Moreover, our previous data showed that severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) open reading frame 3a (ORF3a) and human immunodeficiency virus‐2/SIV viral protein X (Vpx) specifically inhibited cGAS‐STING‐mediated NF‐κB signaling but not IRF3 signaling. 19 , 20 We revealed that HPV18 E7, similar to ORF3a and Vpx, inhibits STING‐triggered NF‐κB activation but not IRF3 activation, whereas KSHV vIRF1 inhibits STING‐triggered IRF3 activation but not NF‐κB activation. These findings are suggestive of a convergent evolution among these viruses toward antagonizing the host's innate immunity. Our results demonstrate that selective suppression of the cGAS‐STING pathway by viral proteins is likely a key pathogenic determinant, making it a promising target for intervention in the development of tumors caused by oncogenic viruses.
2. MATERIALS AND METHODS
2.1. Cell lines and plasmids
HEK293T (CRL‐3216, ATCC) and HeLa cells (CCL‐2, ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin solution. The Myc‐cGAS, STING‐Flag, STING‐Flag truncation, STING‐GFP, KSHV vIRF1, and luciferase reporter plasmids used in this study have already been explored in a previous work. 19 HPV6 E7 (GenBank: QEE83770.1), HPV11 E7 (GenBank: QEE83879.1), and HPV18 E7 (GenBank: ATL15232.1) were synthesized and inserted into the VR1012 vector (Generay Biotech Co., Ltd.). All constructs were confirmed by DNA sequencing.
2.2. Immunoblotting
Immunoblotting was performed as previously described. 19 Specifically, the following antibodies were used: anti‐histone H3 (Genscript, A01502), anti‐HA (BioLegend, 901514), anti‐Flag M2 (Sigma, F3165), anti‐myc (Millipore, 05‐724), anti‐IRF3 (Cell Signaling, 11904), anti‐GAPDH (Proteintech, 60004‐1‐lg), anti‐STING (Proteintech, 19851‐1‐AP), anti‐cGAS (Proteintech, 26416‐1‐AP), anti‐E7 (Santa Cruz, sc‐365035), and anti‐p65 (Proteintech, 10745‐1‐AP). HRP‐conjugated anti‐rabbit IgG (HuaBio, HA1006) and anti‐mouse IgG (HuaBio, HA1001) were used as the secondary antibodies.
2.3. Coimmunoprecipitation
HEK293T cells were seeded into 6‐cm dishes and transfected with the indicated plasmids using Hieff TransTM Liposomal Transfection Reagent (YeasenBiotech, China). Coimmunoprecipitation was performed as previously described. 19
2.4. Nuclear and cytoplasmic fractionation
Nuclear and cytoplasmic protein fractions of the cultured cells were obtained using NE‐PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, 78833) according to the manufacturer's protocols.
2.5. RNA extraction and reverse‐transcription quantitative polymerase chain reaction (RT‐qPCR)
Total RNA was isolated from the cells using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. RNA was reverse‐transcribed using random primers and MultiScribe reverse transcriptase from High‐Capacity cDNA Archive Kits (Applied Biosystems) according to the manufacturer's instructions. The StepOne Real‐Time PCR system (Applied Biosystems) was used for RT‐qPCR amplification. The target sequences have been previously described. 19
2.6. Luciferase assays
HEK293T cells were transfected with the reporter plasmids, Renilla luciferase control plasmid, and the indicated amounts of expression plasmids. Dual‐luciferase activity were measured as previously described. 19
2.7. Immunostaining
Cells grown on coverslips were fixed for 30 min with 4% paraformaldehyde, permeabilized for 10 min in 0.1% TritonX‐100 in phosphate buffered saline (PBS), and blocked with 5% bovine serum albumin for 1 h. Cells were then incubated with the primary antibody at 4°C overnight. After rinsing with PBS, cells were incubated with fluorophore‐conjugated secondary antibodies (MULTI SCIENCES) for 1 h at 37°C. Nuclei were counterstained with DAPI (Sigma‐Aldrich). Images were captured using a laser‐scanning confocal microscope (Zeiss LSM 710) with ZEISS ZEN microscope software used for acquisition.
2.8. Statistical analysis
Data from the luciferase reporter assays and RT‐qPCR results are presented as means and standard errors, and statistical significance was determined using two‐sided unpaired t‐tests (*p < 0.05; **p < 0.01; NS, not significant). All analyses were performed in GraphPad Prism software version 5.01.
3. RESULTS
3.1. HPV18 E7 selectively antagonizes cGAS‐STING‐triggered NF‐κB activation but not IRF3 activation
Using established experimental systems for the detection of cGAS‐STING‐triggered innate immune activation, 19 , 20 we evaluated the innate immune modulation by HPV18 E7 in HEK293T cells. Interestingly, our results showed that HPV18 E7 reduced STING‐induced NF‐κB, NFKBIA, CXCL8, and IFNβ luciferase activity but not IRF3 luciferase activity (Figure 1A,B). We also observed that HPV18 E7 inhibited the expression of STING‐induced NF‐κB‐related genes (NFKBIA, TNFAIP3, CXCL8, and IER3) but not IRF3‐related genes (IFIT1, IFIT2, and IFIT3) (Figure 1C). The level of ectopically expressed cGAS, STING, and HPV18 E7 was physiologically relevant (Supporting Information: Figure 1). Furthermore, NF‐κB luciferase activity and NF‐κB‐related genes were significantly activated by dsDNA in HeLa cells when endogenous HPV18 E7 was silenced (Supporting Information: Figure 2A and B). However, IRF3 luciferase activity and IRF3‐related genes were not activated by dsDNA in these HeLa cells (Supporting Information: Figure 2C−E). We also expressed HPV18 E7 in EA.hy926 cells and evaluated its effects of HPV18 E7 on the DNA‐sensing pathway. As expected, STING agonist‐induced NF‐κB‐related genes, but not IRF3‐related genes, were significantly inhibited in EA.hy926 cells expressing HPV18 E7 compared with that in control cells (Supporting Information: Figure 3). These data indicated that HPV18 E7 selectively antagonizes cGAS‐STING‐triggered NF‐κB activation but not IRF3 activation.
Figure 1.
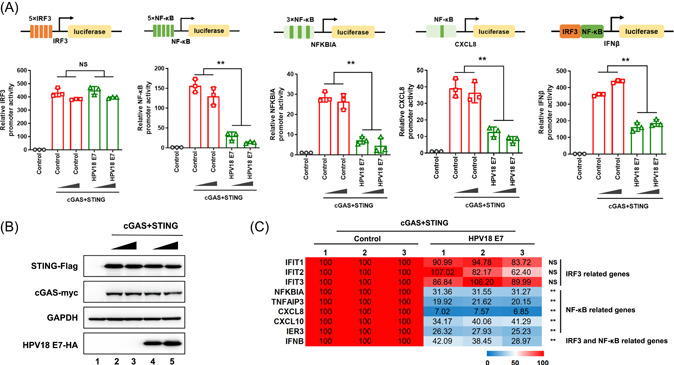
HPV18 E7 selectively antagonizes cGAS‐STING‐triggered NF‐κB activation but not IRF3 activation. (A, B) Schematic diagram of IRF3‐Luc, NF‐κB‐Luc, NFKBIA‐Luc, CXCL8‐Luc, and IFNβ‐Luc constructs. HEK293T cells were transfected with IRF3‐Luc/NF‐κB‐Luc/NFKBIA‐Luc/CXCL8‐Luc/IFNβ‐Luc, pRL‐TK Renilla, Myc‐cGAS and STING‐Flag in the presence or absence of HPV18 E7. (A) Promoter activities and (B) protein expressions were analyzed using a luciferase reporter assay and immunoblotting, respectively. (C) HEK293T cells were transfected with Myc‐cGAS and STING‐Flag in the presence or absence of HPV18 E7. Total mRNA was extracted and analyzed by RT‐qPCR to determine the transcription levels of the indicated genes. Statistical significance was determined using a two‐sided unpaired t‐test (*p < 0.05; **p < 0.01; NS, not significant). The results are shown for N = 3 independent experiments. cGAS‐STING, GMP–AMP synthase‐stimulator of interferon gene; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; RT‐qPCR, reverse‐transcription quantitative polymerase chain reaction.
3.2. HPV18 E7 binds STING in a unique region and blocks the nuclear accumulation of p65 to inhibit NF‐κB signaling
Our previous study showed that deletion of amino acids 344–379 of the STING protein (amino acids 1–343 retained) did not significantly affect cGAS‐STING‐triggered NF‐κB activation but destroyed cGAS‐STING‐triggered IRF3 activation, whereas deletion of amino acids 318–379 of STING (amino acids 1–317 retained) abolished both cGAS‐STING‐triggered NF‐κB and IRF3 activation. 20 (Figure 2A). Because HPV18 E7 can selectively interfere with STING function, we hypothesized that HPV18 E7 can interact with STING. Indeed, an interaction between HPV18 E7 and STING was detected using coimmunoprecipitation (Figure 2B). Interestingly, we discovered that the 318–343 region is required for the interaction of STING with HPV18 E7. By contrast, amino acids 344–379, which are important for IRF3 activation, were dispensable for interaction with HPV18 E7 (Figure 2B). The intracellular co‐localization of HPV18 E7 and STING was observed by immunofluorescence staining of HeLa cells (Figure 2C). Immunoprecipitation analysis using HeLa cells samples indicated an interaction between endogenous STING and HPV18 E7 (Supporting Information: Figure 4). Furthermore, STING activation triggered the nuclear translocation of NF‐κB (p65 and p50) and IRF3. We observed that the STING‐triggered nuclear accumulation of p65 was inhibited by HPV18 E7 (Figure 2D and Supporting Information: Figure 5), consistent with its selective role in inhibiting STING.
Figure 2.
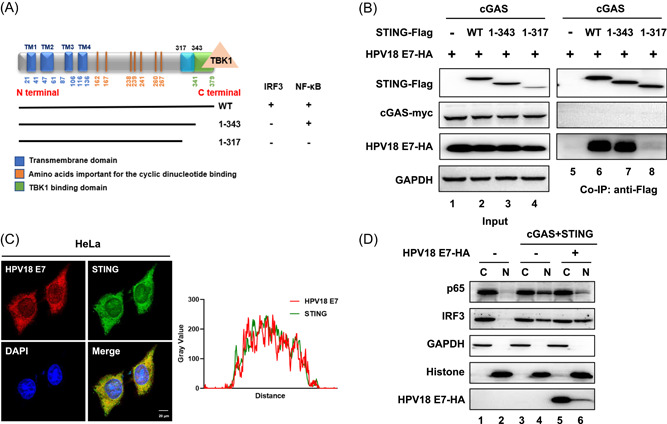
HPV18 E7 binds STING in a unique region and blocks the nuclear accumulation of p65 to inhibit NF‐κB signaling. (A) Schematic of the STING truncation constructs and functions of IRF3/NF‐κB activation. (B) HEK293T cells were transfected with Myc‐cGAS, HPV18 E7 and wild type or truncated STING‐Flag, as indicated. Coimmunoprecipitation analysis was performed as described above. (C) Immunofluorescence staining of endogenous HPV18 E7 and STING in HeLa cells was carried out and followed by laser scanning confocal microscopy. The results were analyzed with ImageJ software to identify colocation of STING and HPV18 E7. (D) HEK293T cells were transfected with Myc‐cGAS and STING‐Flag in the presence or absence of HPV18 E7. The nuclear (N) and cytoplasmic (C) fractions were separated and analyzed using immunoblotting. The results are shown for N = 3 independent experiments. NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; STING, stimulator of interferon gene.
3.3. E7 inhibition of STING‐triggered NF‐κB activation is related to its HPV pathogenicity but not to Rb binding
To investigate the correlation between HPV pathogenicity and cGAS‐STING pathway antagonism by E7, we constructed low pathogenic HPV6 E7 and HPV11 E7 expression vectors. Compared with HPV18 E7, HPV6 E7 and HPV11 E7 did not significantly inhibit STING‐triggered NF‐κB activation (Figure 3A,B). Furthermore, coimmunoprecipitation experiments showed that HPV18 E7, but neither HPV6 E7 nor HPV11 E7, binds to STING (Figure 3C). This indicated that the role of E7 in STING inhibition is related to HPV pathogenicity. High‐risk E7, but not low‐risk E7, interacts with STING and inhibits STING‐triggered NF‐κB activation. Additionally, to evaluate whether the HPV18 E7 LXCXE motif is important for STING‐triggered NF‐κB signaling inhibition, we introduced conservative mutations into this motif (Leu‐Leu‐Cys‐His‐Glu to Val‐Leu‐Ser‐His‐Asp) in HPV18 E7, which is known to disrupt Rb binding (Figure 3D). Interestingly, both wild‐type and mutant HPV18 E7 bound to STING and inhibited STING‐triggered NF‐κB activation (Figure 3E−G). Our findings demonstrate that the mechanism by which HPV18 E7 inhibits the cGAS‐STING pathway is different from that by which HPV18 E7 disrupts the Rb pathway.
Figure 3.
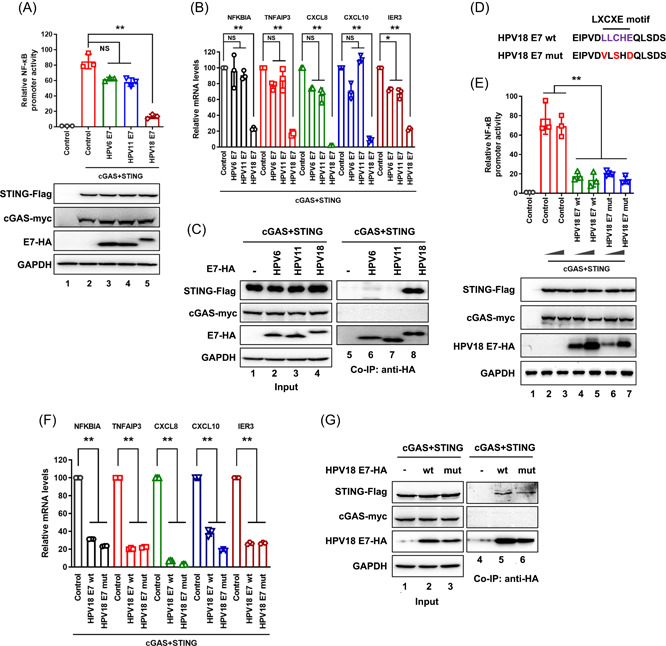
E7 inhibition of STING‐triggered NF‐κB activation is related to its HPV pathogenicity, but not to Rb‐binding. (A) HEK293T cells were transfected with NF‐κB‐Luc, pRL‐TK Renilla, Myc‐cGAS, and STING‐Flag in the presence or absence of different pathogenic HPV E7 plasmids, as indicated. NF‐κB promoter transactivation and protein expression were analyzed. (B–C) HEK293T cells were transfected with Myc‐cGAS and STING‐Flag in the presence or absence of different pathogenic HPV E7 plasmids, as indicated. (B) Total mRNA was extracted and analyzed using RT‐qPCR to determine the transcription levels of the indicated genes. (C) Co‐immunoprecipitation analysis was performed. (D) Sequences of HPV18 E7 containing the LXCXE motif (wild type and mutant). (E) HEK293T cells were transfected with NF‐κB‐Luc, pRL‐TK Renilla, Myc‐cGAS, and STING‐Flag in the presence or absence of HPV18 E7 or its LXCXE motif mutant, as indicated. Promoter activities and protein expression were analyzed using a luciferase reporter assay and immunoblotting, respectively. (F–G) HEK293T cells were transfected with Myc‐cGAS and STING‐Flag in the presence or absence of HPV18 E7 or its LXCXE motif mutant, as indicated. (F) Total mRNA was extracted and analyzed using RT‐qPCR to determine the transcription levels of the indicated genes. (G) Coimmunoprecipitation analysis was performed. Statistical significance was determined using two‐sided unpaired t‐tests (*p < 0.05; **p < 0.01; NS, not significant). The results are shown for N = 3 independent experiments. mRNA, messenger RNA; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; RT‐qPCR, reverse‐transcription quantitative polymerase chain reaction; STING, stimulator of interferon gene.
3.4. Convergent evolution of HPV18 E7, SARS‐CoV‐2 ORF3a, HIV‐2 vpx, and KSHV vIRF1: Selective antagonism of host innate immunity
Our previous studies revealed that SARS‐CoV‐2 ORF3a and HIV‐2/SIV Vpx interact with STING and specifically inhibit cGAS‐STING‐mediated NF‐κB signaling but not IRF3 signaling. 19 , 20 Herein, we identified that HPV18 E7, SARS‐CoV‐2 ORF3a, and HIV‐2 Vpx inhibited STING‐triggered NF‐κB activation but not IRF3 activation. Conversely, KSHV vIRF1 inhibited STING‐triggered IRF3 activation but not NF‐κB activation (Figure 4A−D). It is a universal evolutionary phenomenon that different viral proteins bind to different regions of STING (Figure 4E and Supporting Information: Figure 6) and selectively antagonize STING‐triggered innate immune activation, suggesting convergent evolution of HPV18 E7, SARS‐CoV‐2 ORF3a, HIV‐2 Vpx, and KSHV vIRF1 in antagonizing host innate immunity (Figure 4F).
Figure 4.
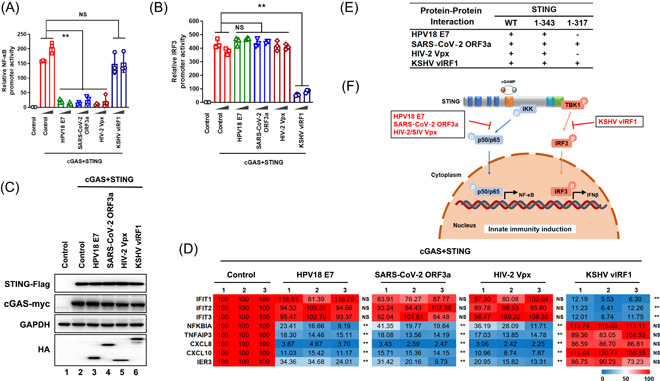
HPV18 E7, SARS‐CoV‐2 ORF3a, HIV‐2 Vpx, and KSHV vIRF1 selectively antagonize cGAS STING‐triggered innate immune activation. (A–C) HEK293T cells were transfected with NF‐κB‐Luc/IRF3‐Luc, pRL‐TK Renilla, Myc‐cGAS, and STING‐Flag in the presence or absence of vectors expressing different viral proteins, as indicated. Promoter activities (A and B) and protein expressions (C) were analyzed using luciferase reporter assays and immunoblotting, respectively. (D) HEK293T cells were transfected with Myc‐cGAS and STING‐Flag in the presence or absence of vectors expressing different viral proteins, as indicated. Total mRNA was extracted and analyzed by RT‐qPCR to determine the transcription levels of the indicated genes. (E) The protein‐protein interactions between different viral proteins and STING wild type or truncated. (F) Schematic representation of cGAS‐STING‐mediated NF‐κB and IRF3 signaling selectively antagonized by different viral proteins. Statistical significance was determined using a two‐sided unpaired t‐test (*p < 0.05; **p < 0.01; NS, not significant). The results are shown for N = 3 independent experiments. HIV‐2, human immunodeficiency virus; mRNA, messenger RNA; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; ORF3a, open reading frame 3a; RT‐qPCR, reverse‐transcription quantitative polymerase chain reaction; STING, stimulator of interferon gene; VPx, Viral protein X.
4. DISCUSSION
Viruses have been central to modern cancer research and have provided profound insights into both the infectious and noninfectious causes of cancer. The cGAS‐STING pathway is an intracellular sensor that activates the interferon pathway in response to viral infection and, thus, mediates the host's defense against a range of DNA and RNA viruses. 5 , 6 However, HPV18 E7 is an antagonist of the cGAS‐STING pathway and interacts with STING to suppress IFNβ production. 15 STING recruits TANK‐binding kinase 1 (TBK1) and IκB kinase (IKK) to trigger downstream IRF3 and NF‐κB activation, respectively. 2 , 3 Herein, we observed that HPV18 E7 selectively antagonizes STING‐triggered NF‐κB activation, but not IRF3 activation, in HEK293T, HeLa and EA.hy926 cells, indicating that IFNβ inhibition is credited to NF‐κB element suppression (Figure 1 and Supporting Information: Figure 1−3). Mechanistically, we revealed that the STING 318–343 amino acid region, which is critical for NF‐κB activation, is required for the interaction between STING and HPV18 E7. HPV18 E7 binds to STING in this region and blocks the nuclear accumulation of p65, but not IRF3, to specifically inhibit NF‐κB signaling (Figure 2 and Supporting Information: Figure 5).
HPV18 is a high‐risk HPV type responsible for most cervical cancers, whereas HPV6 and 11 are prototypical low‐risk types. 7 , 8 , 9 Both high‐ and low‐risk HPV E7 proteins can interact with Rb. 13 Our results indicated that high‐risk E7, but not low‐risk E7, interacts with STING and inhibits STING‐triggered NF‐κB activation (Figure 3). E7 inhibits STING‐triggered NF‐κB activation and is associated with HPV pathogenicity. Moreover, E1A and E7 oncoproteins bind to and inhibit the Rb tumor suppressor pathway via the LXCXE protein motif. 12 Although the E1A LXCXE motif is necessary for antagonizing cGAS‐STING activation, 15 whether the LXCXE motif of E7 is important for E7‐STING interaction remians unknown. We found that both wild‐type and mutant HPV18 E7 oncoproteins inhibited STING‐triggered NF‐κB activation and prevented STING binding (Figure 3), suggesting that the mechanism by which HPV18 E7 inhibits the cGAS‐STING pathway is different from that by which HPV18 E7 disrupts the Rb pathway. A better understanding of the interaction between the DNA tumor virus oncoprotein HPV18 E7 and the cGAS‐STING pathway of human host innate immunity will contribute to the prevention and treatment of HPV‐induced cervical carcinomas.
Broadly, convergent evolution is when two or more organisms from different ancestors evolve similar morphological characteristics or structures as a consequence of similar adaptive pressures. Interestingly, HPV18 E7, SARS‐CoV‐2 ORF3a, and HIV‐2 Vpx inhibited STING‐triggered NF‐κB but not IRF3 activation, whereas KSHV vIRF1 inhibited STING‐triggered IRF3 but not NF‐κB activation, suggesting that different viral proteins bind to different regions of STING and selectively antagonize STING‐triggered innate immune activation (Figure 4). The sequences and evolution of these viral proteins are very different and only distantly related; however, during the long process of viral evolution, the function of antagonizing the cGAS‐STING pathway was selected. Therefore, these viruses appear to have evolved convergently. Consequently, selective antagonism of host innate immunity is a widespread function of DNA viruses, RNA viruses, and retroviruses and has been an important survival skill for viruses throughout evolution.
AUTHOR CONTRIBUTIONS
Meng Lou, Dingbo Huang, and Zhenbang Zhou were the principal investigators of this study. Xiao‐Fang Yu and Meng Lou conceived of and designed the study. Meng Lou and Dingbo Huang performed all experiments together with the assistance of Xinyu Shi, Miaowei Wu, Yajuan Rui, Jiaming Su, and Wenwen Zheng, Meng Lou wrote the manuscript. All authors have read and approved this version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary Figure 1. (A‐C) HEK293T cells were transfected with IRF3‐Luc/NF‐κB‐Luc/NFKBIA‐Luc/CXCL8‐Luc/IFNβ‐Luc, pRL‐TK Renilla, Myc‐cGAS and STING‐Flag in the presence or absence of HPV18 E7. Promoter activities (A) and protein expressions (B and C) were analyzed using a luciferase reporter assay and immunoblotting, respectively. Statistical significance was determined using a two‐sided unpaired t‐test (*P < 0.05; **P < 0.01; NS, not significant). The results are shown for N = 3 independent experiments.
Supplementary Figure 2. (A, B, D) Knockdown HPV18 E7 with siRNA in HeLa cells for 48 h. Then the siNT and siHPV18 E7 cells were transfected with IRF3‐Luc/NF‐κB‐Luc, pRL‐TK Renilla and treated with dsDNA as indicated. Protein expressions (A) and promoter activities (B and D) were analyzed using a luciferase reporter assay and immunoblotting, respectively. (C and E) Knockdown HPV18 E7 with siRNA in HeLa cells. Then HeLa cells were treated with dsDNA as indicated. Total mRNA was exacted and analyzed using RT‐qPCR to determine the transcription levels of the indicated genes. Statistical significance was determined using a two‐sided unpaired t‐test (*P < 0.05; **P < 0.01; NS, not significant). The results are shown for N = 3 independent experiments.
Supplementary Figure 3. (A‐B) EA.hy926 pLVX control cells and pLVX HPV18 E7 stable expression cells were treated with STING agonist (HY‐103665, MCE) for 24 h. The protein expressions were analyzed by immunoblotting (A). Total mRNA was exacted and analyzed using RT‐qPCR to determine the transcription levels of the indicated genes (B). Statistical significance was determined using a two‐sided unpaired t‐test (*P < 0.05; **P < 0.01; NS, not significant). The results are shown for N = 3 independent experiments.
Supplementary Figure 4. The interaction between endogenous STING and HPV18 E7 in HeLa cells was analyzed using immunoprecipitation.
Supplementary Figure 5. (A and B) HEK293T cells were transfected with Myc‐cGAS and STING‐GFP in the presence or absence of HPV18 E7. Immunofluorescent staining was carried out using anti‐p65, anti‐IRF3 and anti‐HA antibodies to detect p65, IRF3 and HPV18 E7, respectively. DAPI staining was performed to show the nucle.
Supplementary Figure 6. HEK293T cells were transfected with Myc‐cGAS, KSHV vIRF1 and wild type or truncated STING‐Flag, as indicated. Co‐immunoprecipitation analysis was performed as described above.
ACKNOWLEDGMENTS
This work was supported in part by funding from the National Natural Science Foundation of China (81802351, 31970151, 92169203, 81701988), National Natural Science Foundation of Zhejiang Province (LY22C080002, LQ21C010001), and Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2019R01007). We thank S. Liu and J. Xuan from the Core Facilities of Zhejiang University School of Medicine for confocal laser scanning microscope assistance. We thank Q. Dong and N. Zhao for providing technical assistance.
Lou M, Huang D, Zhou Z, et al. DNA virus oncoprotein HPV18 E7 selectively antagonizes cGAS‐STING‐triggered innate immune activation. J Med Virol. 2022;95:e28310. 10.1002/jmv.28310
Meng Lou, Dingbo Huang, and Zhenbang Zhou are contributed equally to this study.
DATA AVAILABILITY STATEMENT
All data, materials, and methods are included in the article. The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ng KW, Marshall EA, Bell JC, Lam WL. cGAS‐STING and cancer: dichotomous roles in tumor immunity and development. Trends Immunol. 2018;39(1):44‐54. 10.1016/j.it.2017.07.013 [DOI] [PubMed] [Google Scholar]
- 2. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(2013):786‐791. 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF‐κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci USA. 2021;118(14):e2100225118. 10.1073/pnas.2100225118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674‐678. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phelan T, Little MA, Brady G. Targeting of the cGAS‐STING system by DNA viruses. Biochem Pharmacol. 2020;174:113831. 10.1016/j.bcp.2020.113831 [DOI] [PubMed] [Google Scholar]
- 6. Ma Z, Damania B. The cGAS‐STING defense pathway and its counteraction by viruses. Cell Host Microbe. 2016;19(2):150‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550‐560. 10.1038/nrc2886 [DOI] [PubMed] [Google Scholar]
- 8. Hausen H. Papillomavirus infections: a major cause of human cancers. Biochim Biophys Acta.1996;1288(2):F55‐F78. 10.1016/0304-419X(96)00020-0 [DOI] [PubMed] [Google Scholar]
- 9. Hoppe‐Seyler K, Bossler F, Braun JA, Braun JA, Herrmann AL, Hoppe‐Seyler F, The HPV E6/E7 oncogenes: key factors for viral carcinogenesis and therapeutic targets. TIM. 2018;26(2):158‐168. 10.1016/j.tim.2017.07.007 [DOI] [PubMed] [Google Scholar]
- 10. Dyson N, Howley PM, Münger K, Harlow E. The human papilloma virus‐16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934‐937. 10.1126/science.2537532 [DOI] [PubMed] [Google Scholar]
- 11. McLaughlin‐Drubin ME, Münger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384(2):335‐344. 10.1016/j.virol.2008.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour‐suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391(6670):859‐865. 10.1038/36038 [DOI] [PubMed] [Google Scholar]
- 13. Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8(13):4099‐4105. 10.1002/j.1460-2075.1989.tb08594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moore PS, Chang Y, Why do viruses cause cancer? highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10(12):878‐889. 10.1038/nrc2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS‐STING DNA‐sensing pathway. Science. 2015;350(6260):568‐571. 10.1126/science.aab3291 [DOI] [PubMed] [Google Scholar]
- 16. Lo Cigno I, Calati F, Albertini S, Gariglio M. Subversion of host innate immunity by human papillomavirus oncoproteins. Pathogens. 2020;9(4):292. 10.3390/pathogens9040292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu J, Li W, Shao Y, et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe. 2015;18(3):333‐344. 10.1016/j.chom.2015.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma Z, Jacobs SR, West JA, et al. Modulation of the cGAS‐STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA. 2015;112(31):E4306‐E4315. 10.1073/pnas.1503831112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rui Y, Su J, Shen S, et al. Unique and complementary suppression of cGAS‐STING and RNA sensing‐ triggered innate immune responses by SARS‐CoV‐2 proteins. Signal Transduct Target Ther. 2021;6(1):123. 10.1038/s41392-021-00515-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su J, Rui Y, Lou M, et al. HIV‐2/SIV Vpx targets a novel functional domain of STING to selectively inhibit cGAS–STING‐mediated NF‐κB signalling. Nat Microbiol. 2019;(12):2552‐2564. 10.1038/s41564-019-0585-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. (A‐C) HEK293T cells were transfected with IRF3‐Luc/NF‐κB‐Luc/NFKBIA‐Luc/CXCL8‐Luc/IFNβ‐Luc, pRL‐TK Renilla, Myc‐cGAS and STING‐Flag in the presence or absence of HPV18 E7. Promoter activities (A) and protein expressions (B and C) were analyzed using a luciferase reporter assay and immunoblotting, respectively. Statistical significance was determined using a two‐sided unpaired t‐test (*P < 0.05; **P < 0.01; NS, not significant). The results are shown for N = 3 independent experiments.
Supplementary Figure 2. (A, B, D) Knockdown HPV18 E7 with siRNA in HeLa cells for 48 h. Then the siNT and siHPV18 E7 cells were transfected with IRF3‐Luc/NF‐κB‐Luc, pRL‐TK Renilla and treated with dsDNA as indicated. Protein expressions (A) and promoter activities (B and D) were analyzed using a luciferase reporter assay and immunoblotting, respectively. (C and E) Knockdown HPV18 E7 with siRNA in HeLa cells. Then HeLa cells were treated with dsDNA as indicated. Total mRNA was exacted and analyzed using RT‐qPCR to determine the transcription levels of the indicated genes. Statistical significance was determined using a two‐sided unpaired t‐test (*P < 0.05; **P < 0.01; NS, not significant). The results are shown for N = 3 independent experiments.
Supplementary Figure 3. (A‐B) EA.hy926 pLVX control cells and pLVX HPV18 E7 stable expression cells were treated with STING agonist (HY‐103665, MCE) for 24 h. The protein expressions were analyzed by immunoblotting (A). Total mRNA was exacted and analyzed using RT‐qPCR to determine the transcription levels of the indicated genes (B). Statistical significance was determined using a two‐sided unpaired t‐test (*P < 0.05; **P < 0.01; NS, not significant). The results are shown for N = 3 independent experiments.
Supplementary Figure 4. The interaction between endogenous STING and HPV18 E7 in HeLa cells was analyzed using immunoprecipitation.
Supplementary Figure 5. (A and B) HEK293T cells were transfected with Myc‐cGAS and STING‐GFP in the presence or absence of HPV18 E7. Immunofluorescent staining was carried out using anti‐p65, anti‐IRF3 and anti‐HA antibodies to detect p65, IRF3 and HPV18 E7, respectively. DAPI staining was performed to show the nucle.
Supplementary Figure 6. HEK293T cells were transfected with Myc‐cGAS, KSHV vIRF1 and wild type or truncated STING‐Flag, as indicated. Co‐immunoprecipitation analysis was performed as described above.
Data Availability Statement
All data, materials, and methods are included in the article. The data that support the findings of this study are available from the corresponding author upon reasonable request.