

Abstract
Extracting understanding from the growing “sea” of biological and socioeconomic data is one of the most pressing scientific challenges facing us. Here, we introduce and validate an unsupervised method for extracting the hierarchical organization of complex biological, social, and technological networks. We define an ensemble of hierarchically nested random graphs, which we use to validate the method. We then apply our method to real-world networks, including the air-transportation network, an electronic circuit, an e-mail exchange network, and metabolic networks. Our analysis of model and real networks demonstrates that our method extracts an accurate multiscale representation of a complex system.
Keywords: cellular metabolism, complex networks, multiscale representation
The high-throughput methods available for probing biological samples have drastically increased our ability to gather comprehensive molecular-level information on an ever-growing number of organisms. These data show that these systems are connected through a dense network of nonlinear interactions among its components, and that this interconnectedness is responsible for their efficiency and adaptability. This interconnectedness, however, poses significant challenges to researchers trying to interpret empirical data and to extract the “systems biology” principles that will enable us to build new theories and to make new predictions (1).
A central idea in biology is that life processes are hierarchically organized (2–4). Additionally, it seems plausible that this hierarchical structure plays an important role in the system's dynamics (5). However, given a set of genes, proteins, or metabolites, and their interactions, we still do not have an objective manner to assess whether such hierarchical organization does indeed exist or to identify the different levels in the hierarchy.
Here, we report a method that identifies the levels in the organization of complex systems and extracts the relevant information at each level. To illustrate the potential of our method, it is useful to think of electronic maps such as those provided by Google Maps [see supporting information (SI) Fig. 5]. Electronic maps are powerful tools because they present information in a scalable manner—despite the increase in the amount of information as we “zoom out,” the representation displays the information that is relevant at the new scale. In the same spirit, our method will enable researchers to characterize each scale with the relevant information at that scale. This achievement is key for the development of systems biology, but it will encounter application in many other areas.
Background
Complex networks are convenient representations of the interactions within complex systems (6). Here, we focus on the identification of inclusion hierarchies in complex networks, that is, to the unraveling of the nested organization of the nodes in a network into modules, which in turn are composed of submodules and so on.†
A method for the identification of the hierarchical organization of nodes in a network must fulfill two requirements: (i) It must be accurate for many types of networks, and (ii) it must identify the different levels in the hierarchy as well as the number of modules and their composition at each level. The first condition may appear trivial, but we make it explicit to exclude algorithms that only work for a particular network or family of networks, but that will otherwise fail. The second condition is more restrictive, as it excludes methods whose output is subject to interpretation. Specifically, a method does not fulfill the second condition if it organizes nodes into a tree structure, but it is up to the researcher to find a “sensible” criterion to establish which are the different levels in that tree. An implication of the previous two requirements is that any method for the identification of node organization must have a null output for networks, such as Erdős-Rényi random graphs (10), which do not have an internal structure.
To our knowledge, there is no procedure that enables one to simultaneously assess whether a network is organized in a hierarchical fashion and to identify the different levels in the hierarchy in an unsupervised way. Ravasz et al. (11) studied the hierarchical structure of metabolic networks, but in their analysis the authors put emphasis on detecting “global signatures” of a hierarchical network architecture. Specifically, they reported that for the metabolic networks studied and for certain hierarchical network models the clustering coefficient of nodes appears to scale with the connectivity k as k−1. This scaling, however, is neither a necessary nor a sufficient condition for a network to be hierarchical (12).
More direct methods to investigate the hierarchical organization of the nodes in a network have also been recently proposed (13–15). Although useful in some contexts, these methods do not clearly identify hierarchical levels and thus fail to satisfy condition ii above. Furthermore, all of these methods yield a tree even for networks with no internal structure.
In the following, we define inclusion hierarchies in complex networks and describe an ensemble of hierarchically nested random graphs. We then introduce a method that is able to accurately extract the hierarchical organization of graphs in such an ensemble. Last, we apply our method to several real-world networks.
Inclusion Hierarchies
We start by explicitly defining “networks with a nested hierarchical organization” (see SI Text for a mathematical formulation). We focus on networks that have groups of nodes (modules) that are more densely connected between themselves than they are to other groups of nodes. Each module can in turn have its own internal organization if there are subgroups of nodes within the module (submodules) that are more interconnected than to other nodes in the same module. Note that we assume that the internal organization of each module is solely determined by the connections between the nodes within the module, so that each submodule is completely encapsulated within a larger module.
To test our method, we consider an ensemble of networks, hierarchically nested random graphs, which by construction have a nested hierarchical organization. To construct these networks, we start by defining the nested hierarchy of modules. Suppose we want to create a network with two levels, then we have to define two sets of modules: one at the first level and one at the second level. Consider, for example, a network with 640 nodes that at the first level has four modules comprising 160 nodes each. Each of these four modules will comprise four submodules with 40 nodes each. Once having assigned nodes to groups, we draw an edge between a pair of nodes (i, j) with probabilities (i) p2, if (i, j) are in the same module at the second level; (ii) p1, if (i, j) are in the same module at the first level; and (iii) p0, otherwise. We impose that p2 > p1 > p0, so that the resulting network will have a larger density of connections between nodes grouped in the same submodule at the second level, a smaller density of connections between groups of nodes grouped in the same module at the first level, and an even smaller density of connections between nodes grouped in separate modules at the top level. Thus, the network has by construction a hierarchical organization (see SI Text).
Extracting the Hierarchical Organization of Networks
Our method comprises two major steps (Fig. 1): (i) estimating the “proximity” in the hierarchy between all pairs of nodes, which we call “node affinity”; and (ii) uncovering the overall hierarchical organization of node affinities.
Fig. 1.

Schematic illustration of our method for a simple network. (A) Example network. (B) Modularity landscape. For the example network, there are 15 distinct groupings of nodes into modules. Each large colored circle represents a partition, which we draw inside the circle, with different colors indicating different modules. For clarity, we label each partition with a number from 1 to 15. The color of the partition circle indicates the modularity of that partition following the color code on the bottom right-hand side of the diagram. For simplicity, we consider only single node changes; thus, we connect two partitions, for instance 1 and 2, because the change of a node to a new module in partition 1 generates partition 2. The arrows show the direction of increasing modularity. Local maxima correspond to those partitions that do not point to any other partition; that is, the change of a single node does not increase the modularity. In the example, there are two local maxima: partition 1 and partition 15. To illustrate the concept of basin of attraction, we show next to each partition a colored bar (black and white) that represents the probability that a walker that starts from, for instance, partition 2 and only moves to partitions with larger modularity ends up in either of the local maxima. We use white to indicate partition 15 and black to indicate partition 1. (C) Coclassification matrix. We show the number of times two nodes are classified in the same module, starting from a random partition. Note that nodes a, c and b, d are always classified together because they are in the same module in both local maxima (partitions 1 and 15). In contrast, nodes a and b are only in the same module for one of the maxima (partition 1); therefore, the coclassification is lower than one, but larger than zero. (D) Comparison with randomized networks. In this case, this is the only network that one can build keeping the same degree distribution and not allowing for self-loops. Therefore, the average modularity for the local maxima of the randomized networks and that of the network under analysis are the same. Thus, our conclusion is that this network has no internal organization. (E) Representation of the hierarchical organization for the example network. We show the ordered coclassification matrix on the Left, and on the Right is the tree showing the organization of the nodes into modules. In this case, the network has no significant structure; thus, we show a bar of a single color indicating that there is a single module. Note that a modularity maximization algorithm would have a certain chance (the probability depending on the specific algorithm) of finding partition 15 as the optimal partition and would thus conclude that the network does have a modular structure.
Node Affinity.
A standard approach for quantifying the affinity between a pair of nodes in a network is to measure their “topological overlap” (11, 16, 17), which is defined as the ratio between the number of common neighbors of the two nodes and the minimum degree of the two nodes. This measure identifies affinity between nodes with a dense pattern of local connections. Because topological overlap is a local measure, it will fail to detect any structure when a network is not locally dense (Fig. 2).
Fig. 2.

Affinity measures and clustering methods. (A) We generate a model network comprised of 640 nodes with average degree 16 and with a three-level hierarchical structure (see SI Fig. 8 for results for a network with a “flat” organization of the nodes). We show the affinity matrices Aij obtained for two different measures: (i) topological overlap (11) and (ii) coclassification (see text and Supplementary Information). The color scale goes from red for an affinity of one to dark blue for an affinity of zero. At the far right, we show the hierarchical tree obtained by using two different methods: hierarchical clustering and the “box clustering” method we propose. In the hierarchical clustering tree, the vertical axis shows the average distance, dij = 1 − Aij, of the matrix elements that have already merged. In the box-model clustering tree, each row corresponds to one hierarchical level. Different colors indicate different modules at that level. To better identify which are the submodules at a lower level, we color the nodes in the submodules with shades of the color used for the modules in the level above. Note that topological overlap fails to find any modular structure beyond a locally dense connectivity pattern. In contrast, the coclassification measure clearly reveals the hierarchical organization of the network by the “nested-box” pattern along the diagonal. Significantly, the hierarchical tree obtained via hierarchical clustering fails to reproduce the clear three-level hierarchical structure that the affinity matrix displays, whereas the box-model clustering tree accurately reproduces the three-level hierarchical organization of the network. (B) Accuracy of the method. We generate networks with 640 nodes and with built-in hierarchical structure comprising one (Left), two (Center), and three (Right) levels. The top level always comprises four modules of 160 nodes each. For networks with a second level, each of the top-level modules is organized into four submodules of 40 nodes. For the networks with three levels, each level-two module is further split into four submodules of 10 nodes. We build networks with different degrees of level cohesiveness by tuning a single parameter ρ (see SI Text). For low values of ρ, the levels are very cohesive, for high values of ρ the levels are weakly cohesive. Because we know a priori which are the nodes that should be coclassified at each level, we measure the accuracy as the mutual information between the empirical partition of the nodes and the theoretical one (23). We plot the mutual information versus ρ and, for comparison, we also plot the accuracy of a standard community detection algorithm (24) in finding the top level of the networks (dashed green line). Each point is the average over 10 different realizations of the network. Filled circles, empty squares, and filled diamonds represent the accuracy at the top, middle, and lowest levels, respectively. Note that our method is as good at detecting communities as a standard community detection algorithm for networks with a flat organization of the nodes. Additionally, our method is able to detect the top level for all cases analyzed, whereas standard modularity optimization algorithms are not.
We propose a new affinity measure based on the surveying of the modularity landscape (18), a collective property of the network. Our definition of affinity between nodes draws upon the idea that modules correspond to sets of nodes that are more strongly interconnected than one would expect from chance alone (18).
A partition P of the network is a grouping of nodes into modules, each node belonging to a single module. Let 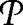 be the ensemble of all partitions of a network into modules (18, 19), and assign to each partition P ∈ the modularity
be the ensemble of all partitions of a network into modules (18, 19), and assign to each partition P ∈ the modularity
 |
where L is the total number of links in the network, li is the number of links within module i, di is the sum of degrees of all of the nodes inside module i, and the sum is over all of the m modules in partition P (Fig. 1A). The modularity of a partition is high when the number of intramodule links is much larger than expected for a random partition.
Currently, researchers believe that the best way to identify densely connected groups of nodes is to find the partition Pop for which M(Pop) is the global maximum of the modularity. However, there are a number of caveats to this idea. First, as illustrated for the very simple network considered in Fig. 1, there may be more than one partition with the maximum value of M(Pop). Second, as demonstrated in refs. 20 and 21, the formulation of M(P) is such that, depending on the density of connections and groups size, it prevents some desired grouping and forces some undesired grouping. Third, Pop may be in practice unreachable if its basin of attraction is too small.
To circumvent these limitations, we propose a different approach. Specifically, we assume that information on the grouping of the nodes into modules is not contained in a single partition but in the entire modularity landscape. In the spirit of the Stillinger and Weber decomposition (22), we propose that the affinity Aij of nodes i and j is determined by those partitions that are local maxima in the landscape and by their basins of attraction.
Let then max be the set of partitions for which the modularity M is a local maxima, that is, partitions for which neither the change of a single node from one module to another nor the merging of two modules will yield a higher modularity (Fig. 1B). The most straightforward way to calculate Aij would be to consider all partitions P̃ ∈ max, and find the fraction for which (i, j) are placed in the same module. However, such a procedure would not take into consideration the size of the basins of attraction of the different maxima. To understand the importance of this fact, consider the “landscape” in Fig. 1 in which each node represents a partition of the network, and for simplicity, we connect two partitions if the change of a single node transforms one partition into the other. This landscape has two local maxima, partitions 1 and 15. Therefore, if we were only to consider those partitions, we would conclude that those partitions are equally important. However, there is no reason to assume that all partitions have the same importance. Actually, for networks with a very clear modular structure, one expects that a few local maxima will yield the most relevant information about the organization of the network. This idea is can be formalized through the concept of basin of attraction.
Consider again the landscape in Fig. 1B. Suppose we wanted to find a partition for which the modularity is a maximum with no a priori information on the landscape. We would start by grouping the nodes into a randomly chosen partition; let us say, partition 13. In partition 13, nodes a and c are placed in one group, whereas nodes b and d are placed into their own groups. There are two single node changes that increase the modularity. Node b can be placed in the same group as node d; this is partition 15, which is a local maxima. Instead, node b can be placed in the same group as nodes a and c; this is partition 14. Partition 14 is not a modularity maximum; thus one would continue our random ascent of the modularity landscape. From partition 14, one could move to partition 1 or to partition 15, both local maxima. This example illustrates that from partition 13, one has a 25% chance of ending in partition 1 and a 75% chance of ending in partition 15. If one repeats this calculation for every possible starting partition, one obtains the size of the basin of attraction of the two local modularity maxima.
Formally, the size of the basin of attraction of P̃ is
 |
where b(P, P̃) is the probability that starting from partition P one ends at partition P̃ ∈ max and ‖‖ is the number of possible partitions (Fig. 1B).
We propose that the affinity Aij of a pair of nodes (i, j) is then the probability that when local maxima partition P̃ ∈ max are sampled with probabilities b(P̃), nodes (i, j) are classified in the same module.
Note that, in contrast to other affinity measures proposed in refs. 9, 15, and 18, the measure we propose does not necessarily coincide with the “optimal” division of nodes into modules, that is, the partition that maximizes M (20). In fact, the modules at the top level of the hierarchy do not necessarily correspond to the best partition found for the global network, even for relatively simple networks (Fig. 2C).
Statistical Significance of the Hierarchical Organization.
Given a set of elements and a matrix of affinities between them, a commonly used tool to cluster the elements and, presumably, uncover their hierarchical organization is hierarchical clustering (25, 26). Hierarchical clustering methods have three major drawbacks: (i) They are only accurate at a local level—at every step a pair of units merge and some details of the affinity matrix are averaged with an inevitable loss of information. (ii) The output is always a hierarchical tree, regardless of whether the system is indeed hierarchically organized or not. (iii) There is no statistically sound general criterion to determine the relevant levels on the hierarchy.
To overcome the first caveat of agglomerative methods such as hierarchical clustering, one necessarily has to follow a top-to-bottom approach that keeps all of the information contained in the affinity matrix. That is the spirit of divisive methods such as k-means or principal component analysis (25), which group nodes into “clusters” given an affinity matrix. However, these methods have a significant limitation: The number of clusters is an external parameter, and, again, there is no sound and general criterion to objectively determine the correct number of clusters.
Because of the caveats of current agglomerative and divisive methods, we propose a “box-clustering” method that iteratively identifies in an unsupervised manner the modules at each level in the hierarchy. Starting from the top level, each iteration corresponds to a different hierarchical level (Fig. 2).
First, to assess whether the network under analysis has an internal organization, we need to compare it with the appropriate null model, which in this case is an ensemble of “equivalent” networks with no internal organization. These equivalent networks must have the same number of nodes and an identical degree sequence. A standard method for generating such networks is the Markov-chain switching algorithm (27, 28). Despite their having no internal structure, these randomized networks have numerous local modularity maxima (19). Thus, to quantify the level of organization of a network, one needs to compare the modularities of the sampled maxima for the original network and its corresponding random ensemble; if the network has a nonrandom internal structure, then local maxima in the original landscape should have significantly larger modularities than local maxima in the landscapes of the randomized networks.
Specifically, for a given network, we compute the average modularity Mav from {M (P̃) : P̃ ∈ max}. Then, we compute the same quantity M avi for each network in the equivalent random ensemble. In virtue of the central limit theorem, the set of average modularities for the whole ensemble {M avi} is normally distributed with mean Mrand and variance σMrand2 (see SI Fig. 6). To quantify the level of organization of a network, we thus compute the z-score of the average modularity z = (Mav − Mrand)/σMrand.
If z is larger than a threshold value zt, then we conclude that the network has internal structure, and we proceed to identify the different modules; otherwise, we conclude that the network has no structure (Fig. 1D). In what follows, we show results for zt = 2.3267, which corresponds to a 1% significance level‡ (SI Text and SI Fig. 9).
Building the Hierarchical Tree.
In networks organized in a hierarchical fashion, nodes that belong to the same module at the bottom level of the hierarchy have greater affinity than nodes that are together at a higher level in the hierarchy. Thus, if a network has a hierarchical organization, one will be able to order the nodes in such a way that groups of nodes with large affinity are close to each other. With such an ordering, the affinity matrix will have a “nested” block-diagonal structure. This is indeed what we find for networks belonging to the ensemble of hierarchically nested random graphs (Fig. 2).
For real-world networks, we do not know a priori which nodes are going to be coclassified together; that is, we do not know which is the ordering of the nodes for which the affinity matrix has a nested block-diagonal structure. To find such an ordering, we use simulated annealing (29) to minimize a cost function that weighs each matrix element with its distance to the diagonal (30)
 |
where N is the order of the affinity matrix (see SI Text and SI Fig. 7). This problem belongs to the general class of quadratic assignment problems (31). Other particular cases of quadratic assignment problems have been suggested to uncover different features of similarity matrices (32). Our algorithm is able to find the proper ordering for the affinity matrix and to accurately reveal the structure of hierarchically nested random graphs (Fig. 2).
The computational cost of this step, the slowest one in our algorithm, limits network sizes to ≈10,000 nodes. However, the cost can be reduced by using faster, but less accurate, methods for ordering the matrix, such as principal component analysis.
Unsupervised Extraction of the Structure.
Given an ordered affinity matrix, the last step is to partition the nodes into modules at each relevant hierarchical level. An ansatz that follows naturally from the considerations in the previous section and the results in Fig. 2 is that, if a module at level ℓ (or the whole network at level 0) has internal modular structure, the corresponding affinity matrix is block-diagonal: At level ℓ, the matrix displays boxes along the diagonal, such that elements inside each box s have an affinity Aℓs, whereas matrix elements outside the boxes have an affinity Bℓ < Aℓs. Note that the number of boxes for each affinity matrix is not fixed; we determine the “best” set of boxes by least-squares fitting of the block-diagonal model to the affinity matrix.
Importantly, we want to balance the ability of the model to accurately describe the data with its parsimony; that is, we do not want to over-fit the data. Thus, we use the Bayesian information criterion to determine the best set of boxes (33).§
To find the modular organization of the nodes at the top level (level 1), we fit the block diagonal model to the global affinity matrix. As we said previously, we assume that the information at different levels in the hierarchy is decoupled, thus to detect submodules beyond the first level, one needs to break the network into the subnetworks defined by each module and apply the same procedure from the start. The algorithm iterates these steps for each identified box until no subnetworks are found to have internal structure.
Method Validation
We validate our method on hierarchically nested random graphs with one, two, and three hierarchical levels. We define the accuracy of the method as the mutual information between the empirical partition and the theoretical one (23). Fig. 2C shows that the algorithm uncovers the correct number of levels in the hierarchy.
Moreover, our method always detects the top level, even for the networks with three hierarchical levels. In contrast, because the partition that globally maximizes M corresponds to the submodules in the second level, even the more accurate module identification algorithms based on modularity maximization would fail to capture the top level organization (20).
The hierarchically nested random graphs considered above have a homogeneous hierarchical structure; however, real-world networks are not likely to be so regular. In particular, for real-world networks, one expects that some modules will have deeper hierarchical structures than others. We thus have verified that our method is also able to correctly uncover the organization of model networks with heterogeneous hierarchical structures (see SI Fig. 10).
Analysis of Real-World Networks
Having validated our method, we next analyze different types of real-world networks for which we have some insight into the network structure: the worldwide air-transportation network (35–37), an e-mail exchange network of a Catalan university (13), and an electronic circuit (4).
In the air-transportation network, nodes correspond to cities (that is, all airports around major cities would be merged into a single node), and two nodes are connected if there is a nonstop flight connecting them. In the e-mail network, nodes are people and two people are connected if they send e-mails to each other. In the electronic network, nodes are transistors and two transistors are connected if the output of one transistor is the input of the other (Table 1).
Table 1.
Top-level structure of real-world networks
| Network | Nodes | Edges | Modules | Main modules |
|---|---|---|---|---|
| Air transportation | 3,618 | 28,284 | 57 | 8 |
| 1,133 | 10,902 | 41 | 8 | |
| Electronic circuit | 516 | 686 | 18 | 11 |
| Escherichia coli KEGG | 739 | 1,369 | 39 | 13 |
| E. coli UCSD | 507 | 947 | 28 | 17 |
We show both the total number of modules and the number of main modules at the top level. Main modules are those composed of more than 1% of the nodes. Note that there is no correlation between the size or number of edges of the network and the number of main modules. KEGG, Kyoto Encyclopedia of Genes and Genomes; UCSD, University of California at San Diego.
We find that the air-transportation network is strongly modular and has a deep hierarchical organization (Fig. 3). This finding does not come as a surprise because historical, economic, political, and geographical constraints shape the topology of the network (35–37). We find eight main modules that closely match major continents and subcontinents and major political divisions, and thus they truly represent the highest level of the hierarchy.¶
Fig. 3.

Hierarchical organization of the air-transportation network. (A) Global-level affinity matrix and hierarchical tree (the representation is the same used in Fig. 2). (B) Top-level modules. Each dot represents a city and different colors represent different modules. Note that the top level in the hierarchy corresponds to major geopolitical units. (C) The “Eurasian” module (which is composed of the majority of European countries, ex-Soviet Union countries, Middle-Eastern countries, India, and countries in Northern half of Africa) splits for levels ℓ = 2 into five submodules. (D) The “Near and Middle East” submodule further splits into seven submodules for ℓ = 3 (D). Note that the air-transportation network is large and very dense (Table 1), and thus the organization of the network is not at all apparent (SI Fig. 11). Remarkably, the modules our method detects show a clear agreement with geopolitical units.
The electronic circuit network is comprised of eight D-flip-flops and 58 logic gates (4). Our method identifies two levels in the network (SI Fig. 12A). At the top level, modules comprise either a D-flip-flop plus some additional gates, or a group of logic gates. At the second level, the majority of modules comprise single gates.
For the e-mail network, five of the seven major modules at the top level (SI Fig. 12B) correspond to schools in the university, with >70% of the nodes in each of those modules affiliated with the corresponding school. The remaining two major modules at the top level are a mixture of schools and administration offices (often collocated on campus), which are distinctly separated at the second level. The second level also identifies major departments and groups within a school, as well as research centers closely related to individual schools.
Application to Metabolic Networks
Finally, we analyze the metabolic networks of E. coli obtained from three different sources‖ (Fig. 4 and SI Fig. 13): the KEGG database (40, 41), the Ma-Zeng database (42), and the reconstruction compiled by Palsson's Systems Biology Laboratory at the UCSD (43). In these networks, nodes are metabolites and two metabolites are connected if there is a reaction that transforms one into the other (44).
Fig. 4.

Hierarchical structure of metabolic networks. (A) Global-level affinity matrices and hierarchical trees for the UCSD reconstruction of the metabolic network of E. coli (43). The overall organization of the network is similar and independent of the reconstruction used to build the network (see SI Fig. 11). (B) We analyze the within-module consistency of metabolite pathway classification for the first (Upper plot) and the second (Lower plot) levels. For each module, we first identify the pathway classifications of the corresponding metabolites; then, we compute the fraction of metabolites that are classified in the most abundant pathway. In the plots, each bar represents one module, its width being proportional to the number of nodes it contains. We color each bar according to its most abundant pathway following the color code on the right-hand side. At the second level (Lower plot), we show each submodule directly below its corresponding top level module. Again, the width of each submodule is proportional to its size. Note that, at the first level (Upper), for all modules except one, the most abundant pathway is composed of more than 50% of the metabolites in the module. Remarkably, at the second level (Lower), for most of the modules all of the metabolites are classified in the same pathway. Moreover, at the second level, we detect smaller pathways that are not visible at the top level.
To quantify the plausibility of our classification scheme, we analyze the within-module consistency of metabolite pathway classification for the top and the second levels of the metabolic network of E. coli (43). For each module, we first identify the pathways represented; then, we compute the fraction of metabolites that are classified in the most abundant pathway. We find that there is a clear correlation between modules and known pathways: At the top level, for all of the modules except one (the central metabolism module), we find that the most abundant pathway comprises >50% of the metabolites in the module.
For the second level, we find that for most of the modules, all of the metabolites are classified in the same pathway. We also detect smaller pathways that are not visible at the top level (such as those for polyketides and nonribosomal peptides, and for secondary metabolites).
Conclusion
Our analysis of model and real-world networks demonstrates that our algorithm is able to provide an objective multiscale description of complex systems that, although not affected by human subjectivity, captures our current understanding of these systems. For example, for the air-transportation network, our method extracts features all the way from continents to country boundaries. Further, our algorithm can be easily generalized to other classes of graphs such as bipartite graphs (e.g., collaboration networks or protein interaction networks obtained from bait–prey data) and directed graphs (e.g., citation networks, food webs, or gene-regulatory networks). The steps of the method would remain unchanged; one would only have to replace Eq. 1 by a suitable measure of modularity for the graph under analysis (45).
Interestingly, for metabolic networks, the algorithm reveals that “known” pathways do not correspond to a single module at the top level, implying that large pathways are in fact composed of smaller units. Intriguingly, these units are not necessarily uniform in “pathway composition” but are a mixture of submodules associated to different pathways. Our results thus prompt the question of how the modules we identify relate to metabolism evolution (46).
More generally, our results have significant implications for systems level approach to the study of cellular processes. A systems approach will only be successful if we are able to develop methods that enable us to extract the small set of information that is significant at the chosen scale of observation, whether this scale is molecular or organismal. A scalable multiscale representation of a biological process, such as the one we demonstrate here, will guide the purposeful design and re-engineering of biological systems for therapeutic purposes.
Supplementary Material
Acknowledgments
We thank U. Alon, A. Arenas, and S. Itzkovitz for providing network data and W. Jiang for advice with the statistical analysis. M.S.-P. and R.G. thank the Fulbright Program and the Spanish Ministry of Education, Culture & Sports. L.A.N.A. gratefully acknowledges the support of the Keck Foundation, the J. S. McDonnell Foundation, and a National Institutes of Health/National Institute of General Medical Sciences K-25 Award.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0703740104/DC1.
We do not consider other hierarchical schemes that classify nodes according to, for instance, their importance (7). Another issue that we do not address here is that of “overlapping” modules. Note also that some authors refer to the existence of “soft” boundaries between communities (8, 9). However, there has been so far no rigorous connection between the soft boundaries and the overlap between communities. Moreover, at present, there is no theoretical model that includes overlapping modules, that is, modules that share nodes, as opposed to communities that share edges.
Results for real networks at a 5% significance level are identical; however, the more stringent threshold is more efficient at detecting the last level in the hierarchy for model networks. Only for a 1–3% of the cases—depending on the cohesiveness of the levels—does the algorithm find one more level than expected.
We have also applied Akaike's information criterion (34), obtaining the same results for nearly all cases.
References
- 1.Pennisi E. Science. 2005;309:94. doi: 10.1126/science.309.5731.94. [DOI] [PubMed] [Google Scholar]
- 2.Oltvai ZN, Barabási A-L. Science. 2002;298:763–764. doi: 10.1126/science.1078563. [DOI] [PubMed] [Google Scholar]
- 3.Alon U. Science. 2003;301:1866–1867. doi: 10.1126/science.1089072. [DOI] [PubMed] [Google Scholar]
- 4.Itzkovitz S, Levitt R, Kashtan N, Milo R, Itzkovitz M, Alon U. Phys Rev E. 2005;71 doi: 10.1103/PhysRevE.71.016127. 016127. [DOI] [PubMed] [Google Scholar]
- 5.Arenas A, Díaz-Guilera A, Pérez-Vicente CJ. Phys Rev Lett. 2006;96:114102. doi: 10.1103/PhysRevLett.96.114102. [DOI] [PubMed] [Google Scholar]
- 6.Amaral LAN, Ottino J. Eur Phys J B. 2004;38:147–162. [Google Scholar]
- 7.Trusina A, Maslov S, Minnhagen P, Sneppen K. Phys Rev Lett. 2004;92:178702. doi: 10.1103/PhysRevLett.92.178702. [DOI] [PubMed] [Google Scholar]
- 8.Reichardt J, Bornholdt S. Phys Rev Lett. 2004;93:218701. doi: 10.1103/PhysRevLett.93.218701. [DOI] [PubMed] [Google Scholar]
- 9.Ziv E, Middendorf M, Wiggins CH. Phys Rev E. 2005;71 doi: 10.1103/PhysRevE.71.046117. 046117. [DOI] [PubMed] [Google Scholar]
- 10.Bollobás B. Random Graphs. 2nd Ed. Cambridge, UK: Cambridge Univ Press; 2001. [Google Scholar]
- 11.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 12.Soffer SN, Vázquez A. Phys Rev E. 2005;71 doi: 10.1103/PhysRevE.71.057101. 057101. [DOI] [PubMed] [Google Scholar]
- 13.Guimerà R, Danon L, Díaz-Guilera A, Giralt F, Arenas A. Phys Rev E. 2003;68 doi: 10.1103/PhysRevE.68.065103. 065103. [DOI] [PubMed] [Google Scholar]
- 14.Clauset A, Moore C, Newman MEJ. Lecture Notes Comput Sci. 2006;4503:1–13. [Google Scholar]
- 15.Pons P. Post-Processing Hierarchical Community Structures: Quality Improvements and Multi-Scale View. 2006 cs.DS/0608050. [Google Scholar]
- 16.Ye Y, Godzik A. Genome Res. 2004;14:343–353. doi: 10.1101/gr.1610504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ihmels J, Bergmann S, Berman J, Barkai N. PLoS Genet. 2005;1:e39. doi: 10.1371/journal.pgen.0010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newman MEJ, Girvan M. Phys Rev E. 2004;69 026113. [Google Scholar]
- 19.Guimerà R, Sales-Pardo M, Amaral LAN. Phys Rev E. 2004;70 doi: 10.1103/PhysRevE.70.025101. 025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fortunato S, Barthélemy M. Proc Natl Acad Sci USA. 2007;104:36–41. doi: 10.1073/pnas.0605965104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumpula JM, Saramaki J, Kaski K, Kertesz J. Eur Phys J B. 2007;56:41–45. [Google Scholar]
- 22.Stillinger FH, Weber TA. Phys Rev A. 1982;25:978–989. [Google Scholar]
- 23.Danon L, Diaz-Guilera A, Duch J, Arenas A. J Stat Mech Theory Exp. 2005:P09008. [Google Scholar]
- 24.Newman MEJ. Proc Natl Acad Sci USA. 2006;103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everitt BS, Landau S, Leese M. Cluster Analysis. London: Arnold; 2001. [Google Scholar]
- 26.Schena M. Microarray Analysis. New York: Wiley; 2002. [Google Scholar]
- 27.Maslov S, Sneppen K. Science. 2002;296:910–913. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]
- 28.Guimerà R, Sales-Pardo M, Amaral LAN. Nat Phys. 2007;3:63–69. doi: 10.1038/nphys489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkpatrick S, Gelatt CD, Vecchi MP. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 30.Wasserman S, Faust K. Social Network Analysis. Cambridge, UK: Cambridge Univ Press; 1994. [Google Scholar]
- 31.Koopmans T, Beckmann M. Econometrica. 1975;25:53–76. [Google Scholar]
- 32.Tsafrir D, Tsafrir I, Ein-Dor L, Zuk O, Notterman DA, Domany E. Bioinformatics. 2005;21:2301–2308. doi: 10.1093/bioinformatics/bti329. [DOI] [PubMed] [Google Scholar]
- 33.Schwarz G. Ann Stat. 1978;6:461–464. [Google Scholar]
- 34.Akaike H. IEEE Trans Automatic Control. 1974;19:716–723. [Google Scholar]
- 35.Barrat A, Barthélemy M, Pastor-Satorras R, Vespignani A. Proc Natl Acad Sci USA. 2004;101:3747–3752. doi: 10.1073/pnas.0400087101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guimerà R, Amaral LAN. Eur Phys J B. 2004;38:381–385. [Google Scholar]
- 37.Guimerà R, Mossa S, Turtschi A, Amaral LAN. Proc Natl Acad Sci USA. 2005;102:7794–7799. doi: 10.1073/pnas.0407994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guimerà R, Amaral LAN. Nature. 2005;433:895–900. doi: 10.1038/nature03288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiele I, Vo TD, Price ND, Palsson BØ. J Bacteriol. 2005;187:5818–5830. doi: 10.1128/JB.187.16.5818-5830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goto S, Nishioka T, Kanehisa M. Bioinformatics. 1998;14:591–599. doi: 10.1093/bioinformatics/14.7.591. [DOI] [PubMed] [Google Scholar]
- 41.Kanehisa M, Goto S. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma H, Zeng A-P. Bioinformatics. 2003;19:270–277. doi: 10.1093/bioinformatics/19.2.270. [DOI] [PubMed] [Google Scholar]
- 43.Reed JL, Vo TD, Schilling CH, Palsson BØ. Genome Biol. 2003;4:R54. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guimerà R, Sales-Pardo M, Amaral LAN. Bioinformatics. 2007;23:1616–1622. doi: 10.1093/bioinformatics/btm150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guimerà R, Sales-Pardo M, Amaral LAN. Phys Rev E. 2007;76 doi: 10.1103/PhysRevE.76.036102. 036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raymond J, Segrè D. Science. 2006;311:1764–1767. doi: 10.1126/science.1118439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}