

Abstract
The cellular amount of proteins not only depends on synthesis but also on degradation. Here, we expand the understanding of differential protein levels by complementing synthesis data with a proteome-wide, mass spectrometry-based stable isotope labeling with amino acids in cell culture analysis of protein degradation in the human pathogen Staphylococcus aureus during glucose starvation. Monitoring protein stability profiles in a wild type and an isogenic clpP protease mutant revealed that 1) proteolysis mainly affected proteins with vegetative functions, anabolic and selected catabolic enzymes, whereas the expression of TCA cycle and gluconeogenesis enzymes increased; 2) most proteins were prone to aggregation in the clpP mutant; 3) the absence of ClpP correlated with protein denaturation and oxidative stress responses, deregulation of virulence factors and a CodY repression. We suggest that degradation of redundant, inactive proteins disintegrated from functional complexes and thereby amenable to proteolytic attack is a fundamental cellular process in all organisms to regain nutrients and guarantee protein homeostasis.
The most essential outcome of bacterial gene expression regulation is that each protein is provided in the appropriate amount at the right time and at the right localization to fulfill its function. On the one hand, the amount of functionally active proteins is determined by the rate of protein biosynthesis on the ribosomes along with subsequent post-translational modifications. On the other hand, stability and structural integrity also have a crucial impact on protein activity. Hence cellular control mechanisms exist to ensure that only intact and functional proteins are preserved at physiologically sufficient amounts and that damaged or redundant proteins are degraded.
Consequently, protein degradation as the final step in the life cycle of a protein is one of the most essential cellular processes to maintain protein homeostasis (1). It is performed by multipartite molecular complexes consisting of chaperones and proteases. In bacteria the Clp proteins constitute the major system to control protein homeostasis. This ATP-dependent molecular degradation machinery is analogous to the eukaryotic 26S proteasome and combines Hsp 100/Clp proteins of the AAA+ superfamily with an associated barrel-like proteolytic chamber (e.g. ClpP). The Hsp 100/Clp proteins are required for unfolding and translocation of substrates to the central proteolytic chamber. Thee highly conserved Clp proteins are involved in cell fitness and stress tolerance in many bacteria including the Gram-positive human pathogen Staphylococcus aureus (2). There are four Clp ATPases (ClpC, ClpX, ClpL, and ClpB) and one Clp protease (ClpP) present in S. aureus and most of them (ClpC, ClpB and ClpP) are regulated by the transcriptional repressor CtsR (3). Because of the emergence of various antibiotic-resistant strains and the concomitant increase in nosocomial infections there is an urgent need for novel antibiotic targets. Because of its high impact on global cellular processes ClpP has attracted attention as such a potential target for novel antibacterial agents (4–6).
Current proteomics technologies allow researchers to monitor bacterial protein stability with a very broad perspective, spanning various levels from single molecule species to the whole proteome. In previous studies we used a two-dimensional gel-based approach to characterize the stability of cytosolic proteins in Bacillus subtilis and S. aureus upon imposition of adverse stimuli such as glucose starvation (7, 8). After pulse labeling with [35S]methionine the remaining radioactivity of electrophoretically separated proteins was monitored during the chase. A gel-based relative quantitation procedure allowed us to assess the stability of single proteins. In starving S. aureus cells many vegetative proteins involved in growth and reproduction were specifically degraded under starvation conditions. These redundant proteins are probably also degraded by Clp proteases in addition to the “classical” Clp substrates such as malfolded, denatured or aggregated proteins. Thus, precursors and energy sources can be made available to the nutrient-starved cell. For instance, the degradation of unemployed ribosomes is probably a huge nutrient reserve during starvation.
The limits of this gel-based pulse-chase labeling technique are identical with the analytical limits of gel-based proteomics (9), only a small portion of the proteome can be resolved on two-dimensional gels. The hydrophobic integral membrane proteins, e.g. totally elude detection by gel electrophoresis. Furthermore, radioactive labeling requires particular safety measures in the laboratory setup and relies on indirect identification by comparison with master gels, which implicates other limitations such as potential mismatches or the dependence on the prior detection by nonradioactive methods. Recently developed highly sensitive and accurate mass spectrometry methods overcome these limitations. In this study, we employed a mass spectrometry-based stable isotope labeling by amino acid in cell culture (SILAC)1 method (10) to simultaneously monitor the accumulation, synthesis and degradation of S. aureus proteins in unprecedented detail. The results reveal a complete picture of the protein degradation patterns in wild type and clpP mutant cells after the transition from a growing to a non-growing state. The methodology can be easily transferred to other pathophysiological conditions such as oxidative stress or iron starvation.
EXPERIMENTAL PROCEDURES
Mutant Construction
For generation of an isogenic clpP mutant the pMAD mutant construction system was used (11). Briefly, a fusion product, which consists of clpP upstream DNA, a spectinomycin resistance marker and clpP downstream DNA (used primers: clpP1-upstream-for 5′-TCCCCCCGGGCAAGTTGAGAGCATTAAATTG-3′; clpP2-upstream-rev 5′-AATTTGTTCGTATGTATTCAACTATATTTCCTCCTTGTAATA-3′; clpP3-downstream-for 5′-TAACAGATTAAAAAAATTATAATTCAAAGTAAAGAGTAGACTAAG-3′; clpP4-downstream-rev 5′-CGGGATCCGGTTACTATCCTCCTGATTTA-3′; spec-fus-for 5′-TTGAATACATACGAACAAATT-3′; spec-fus-rev 5′-TAACAGATTAAAAAAATTATAA-3′) was ligated to the pMAD plasmid for homolog recombination and deletion of clpP in S.aureus COL.
Growth Conditions and Protein Preparation
S. aureus COL cells and the isogenic clpP mutant were grown in CDM (8) containing 0.75 mm amino acid mix with alanine, glycine, valine, cysteine, proline, isoleucine, 2.5 mm glucose and 0.75 mm 13C-lysine, as well as 0.75 mm 13C-arginine for the incorporation of 13C-lysine and 13C-arginine into the proteins (pulse). A 0.03% yeast extract was added in order to allow growth of the clpP mutant as previously described for a Bacillus subtilis clpP mutant (7). When the cells reached the mid-exponential growth phase (optical density at 500 nm of 0.4), they were rapidly transferred to CDM containing 0.75 mm amino acid mix, 1.5 mm glucose, 0.75 mm 12C-lysine, and 0.75 mm 12C-arginine via rapid filtration (chase). This stops the incorporation of heavy lysine and arginine and only allows further incorporation of light lysine and arginine. Samples were taken 20 min after the chase (texp), in the transient phase (ttrans, 0 h) and after 5h (t5 h), 10h (t10 h), 15h (t15 h), 20h (t20 h) of stationary phase. In parallel the cell number was counted by microscopy. Cells were centrifuged (10,000 × g, 4 °C, 5 min) and washed twice with 5 ml TE buffer (10 mm Tris [pH 8.0], 1 mm EDTA) containing 1 mm phenylmethylsulfonyl fluoride. Afterward the pellet was resuspended in 2 ml TE buffer and mechanically disrupted using the Precellys 24 homogenizator (PeqLab, Germany; 4 × 30 s at 6.5 m/s). The glass beads that were used during this procedure were removed via centrifugation (10,000 × g, 4 °C, 5 min) and the supernatant was transferred to a new tube. In a next centrifugation step the cell debris was removed and the supernatant was transferred to an ultracentrifugation tube. The soluble and nonsoluble fraction were separated from the cell lysate by ultracentrifugation (100,000 × g, 4 °C, 1h). The supernatant was decanted as the soluble fraction and the remaining pellet was washed with 3 ml TE buffer and centrifuged again (100,000 × g, 4 °C, 1h). The supernatant was removed and the pellet was solubilized with 70 μl of 10% (w/v) SDS and was considered as the nonsoluble fraction. The whole procedure was performed three times in order to get three biological replicates.
To obtain a 15N-labeled crude protein extract, wild type and clpP mutant cells were grown in 15N-BioExpress 1000 medium either to the midexponential or to the stationary phase. Samples were taken from wild type and clpP mutant via centrifugation (10,000 × g, 4 °C, 5 min). The pellets were pooled and disrupted using the Precellys 24 homogenizator (PeqLab, Germany; 4 × 30 s at 6.5 m/s) to generate a 15N-standard protein extract with labeled proteins from exponential and stationary phase in the wild type as well as in the clpP mutant. The subsequent steps of sample preparation were performed as described above for the pulse-chase samples.
Sample Preparation and Measurement
The protein concentration of each sample was determined via the ninhydrin assay (12). For the GeLC-MS approach the proteins were prefractionated by one-dimensional-SDS-PAGE. 24 μg of the soluble pulse-chase sample were mixed with 7 μg of the 15N-standard and 14 μg of the nonsoluble sample with 14 μg of the nonsoluble 15N-standard followed by a separation with a 12% one-dimensional-PAGE and subsequent tryptic digestion (13). MS and tandem MS (MS/MS) were performed as described previously (14) using an online-coupled LTQ-Orbitrap mass spectrometer (Thermo Fisher).
Data Analysis and Quantification
The entire procedure was adopted from Otto et al. 2010 (14). Spectral data were extracted from the raw measurement files using the Bioworks Browser 3.3.1 SP1 (Thermo Fisher Scientific) with no charge state deconvolution and deisotoping performed on the data to yield *.dta files. Subsequently these data were searched with SEQUEST version v28 (rev.12) (Thermo Fisher Scientific) against a S. aureus COL target-decoy protein sequence database (based on the primary (JCVI) annotation, CMR version 3.0, May 2001) supplemented with a set of common laboratory contaminants compiled with the Bioworks Browser and the reverse sequences of the database to provide an estimate of the false-positive rate of the peptide identification by SEQUEST (15). The searches were performed in three iterations. First, for the GeLC-MS analysis the following search parameters were used: enzyme type, trypsin (K/R); peptide tolerance, 10 ppm; tolerance for fragment ions, 1 amu; b- and y-ion series; variable modification, methionine (15.99 Da); a maximum of three modifications per peptide was allowed. Second iteration: the mass shift of 13C-lysine and 13C-arginine was taken into account in the search parameters. Third iteration: the mass shift of all amino acids completely labeled with 15N-nitrogen was taken into account in the search parameters.
Resulting *.dta and *.out files were merged and filtered using DTASelect (version 2.0.25) (parameters GeLC-MS: -y 2 -c 2 -C 4 -here -decoy rev_ -p 2 -t 2 -u -MC 2 -i 0.3 -fp 0.005). The protein false-positive rate was calculated for each analysis according to Peng et al., 2003 (15). The relative quantification was carried out according to MacCoss et al. 2003 (16). The processed search results were analyzed using the software Census (17) to obtain quantitative data of 12C-peaks (sample), 13C-peaks (sample), 15N-peaks (reference). The ratio of the peak intensities was subsequently calculated for all overview scans contained in the chosen peak boundaries. Peptide ratios were finally exported (peptide determination factor [R2 values] > 0.7, only unique peptides).
Data Processing
First of all, to account for missing data, the quantified peptide raw data were complemented by values obtained by the rule of three (A) 13C/15Nmissing = 12C/15N x 13C/12C, (B) 12C/15 Nmissing = 13C/15N x 12C/13C, and (C) 13C/12Cmissing = 13C/15N x 15N/12C. In a next step we used a hot-deck imputation routine to predict missing values of a peptide based on nonmissing values out of the same data set (18). Therefore we evenly divided the sorted log10 values into 15 classes (quantiles) and calculated the class-specific mean intensity values for each biological replicate. With help of the replicate-specific class membership of the nonmissing peptides we estimated the class-membership of the respective missing peptides. Using the rule of three we then imputed the missing peptide values of a replicate based on the respective class-dependent mean ratios and the nonmissing peptide values of the other replicates. Missing value imputation was only done for peptides with nonmissing values for at least one out of the three replicates.
Afterward quantile-quantile normalization was applied on the replicates and protein values per replicate were calculated as the median of the peptide values if at least two peptides per protein were available. For the calculation of the residual protein amounts (degradation kinetics) the processed 13C/15N values were normalized to the corresponding exponential phase sample and the 13C isotopic protein dilution was corrected by the adjustment to the cell number of each sample. . The linear slope of the log10 values of residual 13C/15N - protein was used to calculate the degradation of each protein.
The isotopic proportions of the soluble and nonsoluble fraction were calculated by normalization against the sum of all isotopic values for each protein in fraction .
Urease Activity Assay
For the urease activity assay cells were grown in tryptic soy broth to an OD540 nm of 2. One milliliter of cells was harvested and the pellet was resuspended in 30 μl of tryptic soy broth followed by the addition of 3 ml urease assay medium (0.1% (w/v) peptone, 0.5% (w/v) NaCl, 0.2% (w/v) KH2PO4, 0.12% (w/v) phenol red, 0.1% (w/v) glucose, 1.8% (w/v) urea), and an incubation of 3 h at 37 °C in a shaker. The urease activity was visualized via color change from orange to red. The test was performed with a control (no cells), wild-type cells and clpP mutant cells.
Intracellular Metabolite Sampling
Samples for intracellular metabolome analysis were obtained by fast vacuum filtration using 0.45 μm filters. Cells were immediately washed on the filter with cold, isotonic NaCl solution (0.6%; <4 °C), subsequently quenched with 60% ethanol (w/v, <4 °C) imbued in liquid nitrogen and stored at −80 °C until further treatment. All following procedures were performed on ice. Defrosted samples were mixed and shaken ten times in alteration to remove cells from the filter. Quenching solution and cells were transferred to tubes filled with glass beads. Cell disruption and simultaneous metabolite extraction was performed by a homogenizer (Precellys 24). Cell debris was removed by centrifugation (8500 rpm, 4 °C, 5 min) and the glass beads were washed with 5 ml distilled water. Afterward the supernatants were pooled and frozen at −80 °C until lyophilization (19).
Intracellular Metabolite Measurement by Ion Pair Liquid Chromatography Coupled Mass Spectrometry (IP-LC/MS)
IP-LC/MS (microTOF) measurements were performed as described previously (20)(Agilent HPLC System 1100; Agilent Technologies, Santa Clara, CA). The injection volume was 25 μl. Chromatographic separation was carried out at room temperature using a RP-C18 Waters® Symmetry Shield column (150 × 4.6 mm, 3.5 μm) with a C18 waters® precolumn. The HPLC was coupled to a micrOTOF (Bruker Daltonics, Bremen, Germany) operating in electrospray negative ionization mode using a mass range from 100 to 2000 m/z. Metabolite amounts were normalized to camphorsulfonic acid (2.5 nmol) as an internal standard.
Intracellular Metabolite Measurement by Gas Chromatography (GC)-MS
GC/MS (quadrupol) analysis was performed using a DB-5 column as described previously (21). Completely lyophilized samples were derivatized for 90 min at 37 °C with MeOX and 30 min at 37 °C with N-methyl-N-(trimethylsilyl) trifluoroacetamide and centrifuged for 2 min at room temperature. The supernatant was transferred into GC vials prior to measurement. Metabolite amounts were normalized to ribitol (20 nmol) as internal standard.
Extracellular Metabolite Sampling and Measurement by 1H-NMR
Two milliliters of cell culture was sterile filtered rapidly and the filtrate was stored at −20 °C. 400 μl of the extracellular sample was buffered to pH 7.0 by addition of 200 μl of a sodium hydrogen phosphate buffer (0.2 m [pH 7.0], including 1 mm TSP) prepared with 50% D2O to provide a nuclear magnetic resonance (NMR) lock signal (22). Spectral referencing was done relative to 1 mm sodium 3-trimethylsilyl-[2,2,3,3-D4]-1-propionic acid (TSP) in phosphate buffer. All NMR spectra were obtained at 600.27 MHz at a nominal temperature of 310 K using a Bruker AVANCE-II 600 NMR spectrometer operated by TOPSPIN 2.1 software (both from Bruker Biospin GmbH, Rheinstetten, Germany). A modified one-dimensional-Nuclear Overhauser enhancement spectroscopy pulse sequence was used with presaturation on the residual HDO signal during both the relaxation delay and the mixing time. A total of 64 free induction decays (FID scans) were collected using a spectral width of 30 ppm for a one-dimensional spectrum (23).
Transmission Electron Microscopy
Cells were fixed with 5% glutaraldehyde in cacodylate buffer 1 (0.2 m cacodylate, 25 mm NaN3; pH 7.0) containing 0.1% ruthenium red for 2 h at room temperature and then at 4 °C over night. Following embedding in low gelling agarose cells were postfixed in 1% osmium tetroxide in cacodylate buffer 2 (0.1 m cacodylate; pH 7.0) containing 0.2% ruthenium red for 2 h at room temperature. After dehydration in graded series of ethanol (30 and 50% ethanol in cacodylate buffer 2 containing 0.2% ruthenium red; 70%, 96%, and 100% ethanol in distilled water) the material was embedded in a mixture of Epon and Spurr (1:2). Sections were cut on an ultramicrotome (Reichert Ultracut, Leica UK Ltd, Milton Keynes, UK), stained with 4% aqueous uranyl acetate for 3 min followed by lead citrate for 30 s and analyzed with a transmission electron microscope LEO 906 (Zeiss, Oberkochen, Germany).
RESULTS
A SILAC-based GeLC-MS Approach Comprehensively Detects the Degradation and Accumulation of S. aureus Proteins in the Soluble and Nonsoluble Proteome Fraction
A SILAC-based GeLC-MS approach (24) was adapted to track the differential protein degradation and synthesis patterns in a S. aureus wild type and an isogenic clpP mutant strain during exponential, transient and glucose starvation-induced stationary growth phase on a proteome-wide scale (Fig. 1). The proteins were labeled in a chemically defined medium containing the heavy amino acids 13C-arginine and 13C-lysine. When the cells reached the mid-exponential growth phase they were rapidly shifted to a medium containing light amino acids for a chase (12C-arginine, 12C-lysine) followed by sampling 20 min after the chase in the exponential phase, in the transient phase as well as 5 h, 10 h, 15 h, and 20 h after entry into stationary phase. To quantify the single isotopic 13C- and 12C-protein species an external 15N-standard was added to each sample. The standard was obtained by pooling wild type and mutant cells from exponential and stationary growth phase cultivated in 15N-supplemented medium. Soluble cytosolic proteins were separated from nonsoluble or aggregated proteins by ultracentrifugation (14, 25). All in all, ∼900 proteins were detected and quantified. The high validity and reproducibility of this approach is substantiated by low coefficients of variation of the protein quantitation data (supplemental Fig. S1A–S1D). The kinetics of protein degradation in glucose-starved cells could be monitored by assessing the decrease of the heavy 13C proteins. Conversely, the quantitation of proteins synthesized after the shift into the light 12C medium reveals the rate of de novo synthesis as well their protein degradation during the chase. The sum of the 13C and 12C measurements of a protein in a sample reflects the total accumulated amount of that protein. Furthermore, the inclusion of the non-soluble protein fraction into the analysis yielded valuable insight into the dynamics of protein denaturation and aggregation. In total, the extensive data set from this study contains comprehensive information on the life cycle of each labeled and detected protein and at the same time reveals similar life cycle patterns within groups of physiologically and functionally correlated proteins.
Fig. 1.

Schematic presentation of the experimental procedure and the possible events that can be observed. A, protein degradation; B, protein stabilization; C, protein de novo synthesis; D, protein shift from the soluble into the non-soluble cell fraction; E, accumulation of 13C and 12C species protein species.
Protein Degradation During Starvation Differentially Affects Proteins Needed for Growth
The quantitative 13C profile directly indicates the degree of (in)stability. Approximately 250 of the 900 detected proteins showed a decrease in quantity to 50% or less during the time course. Many of these degraded proteins required for growth appear to lack a functional relevance under glucose starvation. Voronoi treemaps were used for better visualization (26). In these treemaps proteins can be visually clustered according to specific qualifiers, e.g. into functional categories (Fig. 2) or regulatory groups (supplemental Fig. S2B). A timeline series of treemaps along the growth curve depicts both single profiles and general trends (Fig. 2, supplemental Fig. S2). Although the visual stratification by common regulators (supplemental Fig. S2B) shows an irregular, fuzzy general picture, the spatial representation on the basis of functional categories reveals hot spots of preferentially degraded proteins (Fig. 2, supplemental Fig. S2A).
Fig. 2.

Treemaps visualize an increase and a decrease of proteins in the wild type and clpP mutant for the soluble as well as the nonsoluble fraction. The linear slope was calculated for each protein out of the log10 13C/15N - values corrected against isotopic protein dilution because of growth and normalized against the initial 13C/15N-value for each fraction . Proteins were clustered according to their functional categories (e.g. protein synthesis). Orange colors designate a decrease, blue colors an increase and whitish colors stable proteins. Gray polygons indicate undetermined proteins.
The mainly affected categories comprise (1) proteins with vegetative functions for growth and reproduction (e.g. ribosomal proteins), (2) anabolic enzymes and (3) selected catabolic enzymes.
(1) Proteins required for growth and reproduction were rapidly degraded in nongrowing cells, particularly ribosomal proteins (e.g. RplA, RplQ, RpsJ, and RpsK), but also other proteins involved in translation (e.g. RluB, TrmU), cell wall synthesis (DltC, GltB) and other vegetative processes (e.g. Pls, UreG) (supplemental Fig. S3). As previously suggested (9) and confirmed in this study, the majority of ribosomal proteins were degraded in nongrowing cells (Fig. 2). To our surprise the abundant translational factors such as Ef-Tu and Ef-Ts and aminoacyl-tRNA synthetases remained relatively stable.
(2) Most of the anabolic enzymes that are required no longer or in lower amounts in starving cells were degraded, but with different kinetics. Pnp and NrdEF were rapidly proteolyzed, whereas other enzymes involved in purine and pyrimidine nucleotide synthesis decreased less strongly (supplemental Fig. S4C). Enzymes responsible for amino acid synthesis were also degraded to different extents (supplemental Fig. S4D, S4E). Although LeuCD, IlvB, and IlvA2 were promptly broken down, other enzymes were degraded at a smaller rate or remained stable (e.g. LeuB).
(3) Different results were obtained for catabolic enzymes dependent on their requirements in glucose-starved cells. After glucose exhaustion the level of glycolytic enzymes (Fig. 3, supplemental Fig. S4A) diminished, and all pentose phosphate pathway enzymes appeared slightly degraded (supplemental Fig. S4B). In contrast, TCA cycle enzymes remained stable in the stationary growth phase and, moreover, were extensively synthesized in glucose-starved cells (supplemental Fig. S5).
Fig. 3.

Degradation kinetics of selected proteins involved in glycolysis and TCC depicted as heatmap for soluble and nonsoluble fraction (13C/15N - values corrected against isotopic protein dilution because of growth and normalized against the initial 13C/15N-value for each fraction; . Glycolytic enzymes were slightly degraded, whereas most of the TCA cycle enzymes were stabilized or shifted into the nonsoluble fraction (e.g.. PdhABCD).
In addition, several new proteolytic targets (supplemental Table S1) were identified, such as the pathogenicity factors SarR and SarZ (supplemental Fig. S3).
Some Proteins Shift to the Nonsoluble Fraction in the Wild Type
In order to follow the fate of proteins prone to degradation, the nonsoluble protein fraction was also analyzed. Malfolded, (partially) denatured proteins were enriched in this fraction before degradation by proteolytic systems or, in some cases, before renaturation by chaperones. A few proteins shifted from the soluble to the nonsoluble fraction and remained relatively stable in that fraction (Fig. 2). This transition was particularly pronounced for the complete pyruvate dehydrogenase complex (PdhABCD); IlvC, IlvD, and ThrC; SerA as well as PrsA (Figs. 3, 4, supplemental Figs. S4A, S4C, S4E). However, for most proteins the shift toward the nonsoluble fraction is perfunctory in the wild type. Furthermore, numerous degradation candidates could not be detected in the nonsoluble fraction at all (Fig. 2, supplemental Fig. S2A), indicating that proteolysis of aggregated proteins is a rapid process or soluble proteins can be targeted to proteolysis without an aggregated intermediate form.
Fig. 4.

The proportion of 13C and 12C isotopic forms of selected proteins is depicted for the wild type and the clpP mutant in the soluble and nonsoluble fraction; (exp. growth phase, transient phase (0 h), 5 h, 10 h, 15 h, and 20 h of stationary phase). The isotopic proportions of the soluble and nonsoluble fraction were calculated by normalization against the sum of all isotopic values for each protein in a fraction .
The Phenotype of a clpP Deletion Mutant Strongly Deviates From the Wild Type
To further characterize cellular protein degradation on a proteome-wide scale, we constructed an in-frame deletion mutant of the major cellular protease, ClpP. Previous data revealed that ClpP is necessary for a variety of cellular processes. The protease appeared essential for growth at low and high temperatures, virulence, and autolysis (27–29). The growth curve illustrates the retarded growth under normal conditions as compared with the wild type (supplemental Fig. S6). The mutant grew with a much longer generation time (gwild type = 1.16 h; gΔclpP = 2.03 h) and did not reach the same final optical density as the wild type. In addition, the mutant showed higher urease activity as described earlier (27), stronger staphyloxanthin pigmentation and a thicker cell wall than the wild type (supplemental Figs. S6, S7). The intense pigmentation may indicate oxidative stress in the clpP mutant and probably represents an important defense mechanism against reactive oxygen species (ROS) (30).
Proteins are Prone to Aggregation in the clpP Mutant
The 13C-labeled proteins continuously declined in the soluble fraction of both the wild type and the clpP mutant strain (Fig. 5). On the other hand, the ΔclpP strain showed a dramatically increased level of 13C-labeled proteins in the non-soluble fraction in the stationary phase (Figs. 2, 5, supplemental Fig. S2). Massive protein aggregation occurs, possibly owing to the diminished degradation capacity and energy depletion.
Fig. 5.

Loess-fit curves over all 13C-remaining protein values (13C/15N - values corrected against isotopic protein dilution because of growth and normalized against the initial 13C/15N-value for each fraction; reveal the protein stability in the soluble (blue) and in the nonsoluble fraction (red) of the wild type and the clpP mutant. The different reds and blues correspond to the three biological replicates.
The Absence of ClpP Correlates with Protein Denaturation and Oxidative Stress Responses as well as with Deregulation of Virulence Factors
The renaturation process of aggregated proteins in the clpP mutant coincided with a derepression of the CtsR regulon as a result of protein stress (3). The higher levels of ClpC, ClpB, GroEL, DnaK, DnaJ, and GrpE and the absence of ClpP (Fig. 6, supplemental Figs. S8, S9) probably promoted the renaturation of damaged and aggregated proteins. Stronger protein aggregation in the clpP mutant could be detected for several proteins (e.g. NrdEF, Pnp, SufB, and ribosomal proteins, Figs. 2, 4, supplemental Fig. S3). Interestingly, the clpP mutant faced severe oxidative stress as implied by the induction of numerous members of the PerR regulon (Fig. 6, supplemental Fig. S9).
Fig. 6.

A, A Voronoi treemap of clustered regulons. Values reflect protein accumulation ratios from the wild type and the clpP mutant during transient growth. ± indicates positive or negative control by the regulator. B, The protein accumulation ratios (log10) from selected proteins are depicted for the CodY-, CtsR-, HrcA-, Fur-, PerR-, and LexA-regulon. Blue indicates stronger accumulation in the wild type and dark orange indicates stronger accumulation in the clpP mutant.
In addition, the lack of ClpP affected the amount of some virulence-associated regulators (supplemental Fig. S10). For example, SarS was increased in the soluble and non-soluble fraction and SarA only in the non-soluble fraction. SrrA was down-regulated in the ΔclpP strain.
The Reduced Enzyme Levels for Branched Chain Amino Acid and Purine Nucleotide Biosynthesis and a High GTP Level Indicate a CodY Repression in the clpP Mutant
One of the most striking observations was that the level of enzymes involved in branched-chain amino acid biosynthesis (e.g. LeuA, LeuB, LeuC, LeuD; IlvA2, IlvB, IlvC, and IlvD) and purine nucleotide biosynthesis (e.g. PurC, PurD, PurE, PurL, PurM, PurQ, and PurS) were drastically down-regulated in the clpP mutant (Fig. 6, supplemental Fig. S11). To determine the cause of this effect, the extra- and intracellular metabolite pool was measured (supplemental Fig. S12). As expected for glucose-starved cells, glucose and glycine were rapidly consumed by the wild type and the clpP mutant at a similar rate; however, alanine, isoleucine and proline were not taken up by the clpP mutant from the extracellular medium. Although branched-chain amino acids had lower absolute intracellular concentrations in the clpP mutant than in the wild type (supplemental Fig. S12), the relative amount of e.g. isoleucine when normalized to the amount of CodY was roughly the same (Fig. 7). Besides branched-chain amino acids, GTP is an even more potent effector molecule of the auto-regulated CodY regulator (31, 32). The normalized GTP level was increased in the ΔclpP strain as compared with the wild type, which may render the CodY repressor more active in the mutant (Fig. 7).
Fig. 7.
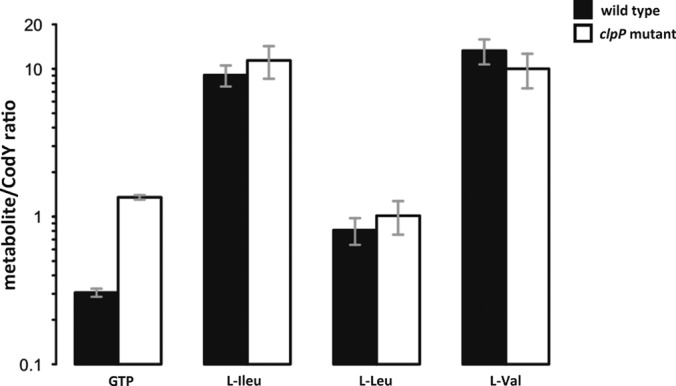
The amount of intracellular GTP, l-isoleucine, l-leucine, and l-valine in the exponential growth phase was normalized to the total amount of CodY protein.
The GeLC-MS Results are Consistent with Previous Two-dimensional-PAGE Analyses
A set of significantly unstable proteins was compared with previously identified degradation candidates detected in a two-dimensional-PAGE pulse-chase approach (Fig. 8, supplemental Fig. S13) (8). Apart from a remarkable overlap of degraded proteins between the two studies, ∼200 new protein degradation candidates could be exclusively retrieved by the GeLC-MS approach. The main degradation candidates such as NrdE, NrdF, LeuD, LeuC, TpiA, and Pnp were identified as degraded during glucose starvation in wild-type cells with both experimental setups (Fig. 4, supplemental Figs. S3, S13). However, some previously described proteolytic substrates (e.g. LeuA, LeuB, MetS, CysS, and SucB) appeared stable in the mass-spectrometry approach probably because of a higher reproducibility of the methodology. An additional advantage of the GeLC-MS analysis was the sensitive and reliable detection of proteins that are refractory to separation by two-dimensional gel electrophoresis such as aggregated and other physico-chemically “challenging” proteins with e.g. an extreme pI, molecular weight or high hydrophobicity (9, 33, 34).
Fig. 8.

A Treemap is shown for the comparison of results obtained by two-dimensional-PAGE and mass spectrometry. Proteins detected in the soluble fraction in the wild type are colored in light gray; proteins solely found by mass spectrometry to be diminished below 50% are colored in red; proteins solely detected by two-dimensional-PAGE to be diminished below 50% are colored in blue; proteins with a decrease below 50% detected by both methods are colored white.
DISCUSSION
A pulse-chase SILAC (24) and a GeLC-MS approach were employed for the integrated investigation of protein degradation, stability and accumulation. They combined tracking the amount of single proteins synthesized at certain growth stages and in different protein fractions on a global scale. Previously applied experimental methods were mainly based on radioactive pulse-chase labeling and two-dimensional-PAGE or immunoprecipitation followed by one-dimensional-PAGE (8, 28, 35, 36). Immunoprecipitation has the advantage of reliable quantitative results for a specific protein, but only for one or a few protein(s) of interest against which antisera have been raised. The radioactive pulse-chase approach coupled with two-dimensional-PAGE can simultaneously monitor several hundred proteins (9), yet these proteins only represent a limited, analytically accessible portion of the proteome owing to physico-chemical properties (e.g. pI 4–7, alkaline or nonsoluble proteins). Besides, two or more proteins may appear as only one spot (34, 37), severely complicating the data analysis for these proteins. The SILAC and GeLC-MS methods in this study couple the strength of direct protein identification via MS/MS spectra with the power to reliably quantify hundreds of proteins (9) (supplemental Fig. S1). All in all, ∼1,400 proteins could be identified and the kinetic data of 900 different proteins were obtained over the time course. As compared with previous work, we were able to verify most of the protein degradation candidates found by two-dimensional-PAGE analyses and revealed additional degradation candidates (Fig. 8, supplemental Table S1). Nevertheless, there were also some drawbacks, such as missing quantitation values for certain proteins at time points for which no respective peptides could be detected.
Our data show that vegetative proteins of S. aureus involved in growth and reproduction, e.g. ribosomal, translation and cell wall/membrane synthesis proteins were degraded during glucose starvation in the wild type, but remained stable in the clpP mutant. Similarly, selected groups of anabolic (e.g. Pnp, NrdEF, LeuCD, IlvB, and IlvA2) and catabolic enzymes (e.g. for glycolysis) were affected by degradation. This observation suggests that proteins,which are no longer required, disintegrate from functional multicomponent complexes and convert into degradation substrates of the Clp proteolytic machinery. According to this hypothesis active proteins are protected against degradation by incorporation into functional complexes. We suggest that the formation of these complexes relies on hydrophobic protein-protein interaction surfaces. When the metabolic throughput falls below a critical threshold the complexes become idle and decompose so that hydrophobic regions of the respective enzymes are recognized by the degradation machinery.
Because the data-set of this study includes the quantitation of 12C-labeled proteins we could also discern stable proteins that were continuously synthesized or only synthesized and accumulated in glucose-starved cells. The most prominent examples are enzymes catalyzing reactions for gluconeogenesis (e.g. GapA2, PckA) and in the TCA cycle (e.g. GltA, AcnA, SucB, FumC) (supplemental Fig. S14). This observation supports the rationale that metabolic capacities are tightly adjusted to the prevailing physiological conditions. In this instance, the “reinforcement” of gluconeogenesis and the TCA cycle counteract the deprivation of glucose as a preferred carbon and energy source. Under glucose starvation the TCA cycle is the main source for NADH generation and ATP synthesis. The TCA cycle is fed by degradation products of proteins, fatty acids etc. and by carbon overflow products such as acetate (22), obviously the main carbon sources in glucose-starved cells.
Aggregation of bacterial cytoplasmic proteins was reported previously (38–40). We also observed protein shifts from the soluble to the non-soluble fraction during the entry into stationary phase. For example, the entire pyruvate dehydrogenase complex (PdhABCD) and other enzymes (SdhA and SdhB; IlvC, IlvD and ThrC; SerA as well as PrsA) aggregated. It is unclear whether this shift has a physiological relevance in terms of a targeted metabolic control or whether it is primarily linked to physico-chemical parameters such as molecular weight or hydrophobicity.
However, in cells lacking the protease ClpP the level of aggregated proteins was drastically increased, corresponding with distinct phenotypic differences from the wild type (e.g. reduced growth rate, higher urease activity, thicker cell wall, stronger pigmentation). This underlines the high physiological importance of ClpP for protein homeostasis also in S. aureus. As judged by the increasing 13C-level in the non-soluble fraction of the clpP mutant at later time points, the degree of aggregation remained high throughout the time course. Some proteins yet seemed to be unaffected in their stability profile without ClpP. For example, the ribosomal protein RpsG diminished rapidly both in the wild type and the mutant strain. Because no Lon protease is found in Staphylococci, the proteases FtsH, SACOL0369 (putative paralog of clpP) or ClpYQ (HslUV) could be responsible for ClpP-independent protein degradation.
The clpP mutant also showed clear symptoms of protein denaturation and oxidative stress responses. In the early stages of protein aggregation reactive oxygen species occur (41–43), which induced PerR-regulated oxidative stress proteins such as AhpCF (alkyl hydroperoxidereductase) and FtnA (ferritin) for detoxification. Furthermore a strong SOS response in the clpP mutant (27) indicated DNA damage (44). The strong protein stress in the clpP mutant also correlated with a derepression of the CtsR-regulon (45) resulting in increased chaperone levels. Until entry into stationary phase the ATP level might be sufficiently high (46, 47) to provide energy for the ATP-dependent renaturation of aggregated proteins by the chaperones occurring in the clpP mutant only but not in the wild type. Later, glucose depletion leads to a drop in the ATP level (22) and the renaturation process is reduced.
GTP and l-isoleucine are effector molecules of the CodY repressor, a central regulator of biochemical pathways involved in nitrogen utilization. High levels of these effector molecules cause CodY activation and a strong repression of CodY-dependent genes (31, 48). Proteins encoded by the pur-operon (purEKCSQLFMNHD), the glt-operon (gltBD), the suc-operon (sucAB), the pyc gene, the opp-operon (oppBCDF), and the codY-operon (xerC-hslUV-codY) were found strongly decreased in their amount and synthesis in the clpP mutant. According to our results, a higher GTP level seems to be particularly relevant for CodY repression in the ΔclpP strain.
Over the past years the physiological role of ClpP was extensively studied (27, 49–52). Because a clpP mutant possesses a low virulence potential (27, 52, 53) this protease seems to be a promising target for novel antibiotic agents against S. aureus (54, 55). Interestingly, the global virulence regulator SarA was constitutively expressed, but massively aggregated in the clpP mutant. Furthermore, SarS was present at a higher amount in the clpP mutant suggesting a modified or nonfunctional SarA protein, which could no longer repress sarS (56, 57).
In summary, we were able to globally monitor protein synthesis, accumulation, degradation and shifts between the soluble and non-soluble fraction with high resolution and reproducibility and propose a working model for a clpP mutant in S. aureus (supplemental Fig. S15). We found that proteolysis in S. aureus is highly selective (supplemental Fig. S16) as well as dependent on ClpP for most proteins and that protein stability both of single and of functionally related groups of proteins is closely linked to the current physiological requirements and restrictions. We suggest that proteolysis of “unemployed,” disintegrated and inactive proteins is a fundamental process of cellular regulation in all organisms to regain nutrients and to guarantee protein homeostasis. This study establishes the basis for further work on the dynamic balance between protein synthesis, degradation and accumulation in S. aureus and other bacteria under various growth and stress conditions.
Supplementary Material
Acknowledgments
We thank Alexander Elsholz for critical reading of the manuscript and helpful suggestions and Mathias Berth and Frank Schmidt for the discussion concerning the biomathematics.
Footnotes
* This work was supported by grants of the Deutsche Forschungsgemeinschaft (SFB-TRR34), Bundesministerium für Bildung und Forschung (ZIK FunGene 03Z1CN21), and European Union (Basysbio LSHG-CT-2006-037469) to M.H.
 This article contains supplemental Fig. S1.
This article contains supplemental Fig. S1.
1 The abbreviations used are:
- SILAC
- stable isotope labeling by amino acid in cell culture
- MS/MS
- tandem MS.
REFERENCES
- 1. Mogk A., Huber D., Bukau B.(2011) Integrating protein homeostasis strategies in prokaryotes. Cold Spring Harb. Perspect. Biol. 3, a004366-a004366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frees D., Chastanet A., Qazi S., Sørensen K., Hill P., Msadek T., Ingmer H. (2004) Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol. Microbiol. 54, 1445–1462 [DOI] [PubMed] [Google Scholar]
- 3. Elsholz A. K., Gerth U., Hecker M. (2010) Regulation of CtsR activity in low GC, Gram+ bacteria. Adv. Microb. Physiol. 57, 119–144 [DOI] [PubMed] [Google Scholar]
- 4. Staub I., Sieber S. A. (2008) Beta-lactams as selective chemical probes for the in vivo labeling of bacterial enzymes involved in cell wall biosynthesis, antibiotic resistance, and virulence. J. Am. Chem. Soc. 130, 13400–13409 [DOI] [PubMed] [Google Scholar]
- 5. Sass P., Josten M., Famulla K., Schiffer G., Sahl H.-G., Hamoen L., Brötz-Oesterhelt H. (2011) Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc. Natl. Acad. Sci. U.S.A. 108, 17474–17479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brötz-Oesterhelt H., Beyer D., Kroll H. P., Endermann R., Ladel C., Schroeder W., Hinzen B., Raddatz S., Paulsen H., Henninger K., Bandow J. E., Sahl H. G., Labischinski H. (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 7. Gerth U., Kock H., Kusters I., Michalik S., Switzer R. L., Hecker M. (2008) Clp-dependent proteolysis down-regulates central metabolic pathways in glucose-starved Bacillus subtilis. J. Bacteriol. 190, 321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michalik S., Liebeke M., Zühlke D., Lalk M., Bernhardt J., Gerth U., Hecker M. (2009) Proteolysis during long-term glucose starvation in Staphylococcus aureus COL. Proteomics 9, 4468–4477 [DOI] [PubMed] [Google Scholar]
- 9. Becher D., Hempel K., Sievers S., Zühlke D., Pané-Farré J., Otto A., Fuchs S., Albrecht D., Bernhardt J., Engelmann S., Völker U., van Dijl J. M., Hecker M. (2009) A proteomic view of an important human pathogen - towards the quantification of the entire Staphylococcus aureus proteome. PLoS ONE 4, e8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doherty M. K., Hammond D. E., Clague M. J., Gaskell S. J., Beynon R. J. (2009) Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J. Proteome Res. 8, 104–112 [DOI] [PubMed] [Google Scholar]
- 11. Arnaud M., Chastanet A., Débarbouillé M. (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starcher B. (2001) A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal. Biochem. 292, 125–129 [DOI] [PubMed] [Google Scholar]
- 13. Dreisbach A., Otto A., Becher D., Hammer E., Teumer A., Gouw J. W., Hecker M., Völker U. (2008) Monitoring of changes in the membrane proteome during stationary phase adaptation of Bacillus subtilis using in vivo labeling techniques. Proteomics 8, 2062–2076 [DOI] [PubMed] [Google Scholar]
- 14. Otto A., Bernhardt J., Meyer H., Schaffer M., Herbst F. A., Siebourg J., Mäder U., Lalk M., Hecker M., Becher D. (2010) Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nat. Commun. 1, article 137, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 16. MacCoss M. J., Wu C. C., Liu H., Sadygov R., Yates J. R., 3rd (2003) A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal. Chem. 75, 6912–6921 [DOI] [PubMed] [Google Scholar]
- 17. Park S. K., Venable J. D., Xu T., Yates J. R., 3rd (2008) A quantitative analysis software tool for mass spectrometry–based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aittokallio T. (2010) Dealing with missing values in large-scale studies: microarray data imputation and beyond. Brief Bioinformatics 11, 253–264 [DOI] [PubMed] [Google Scholar]
- 19. Meyer H., Liebeke M., Lalk M. (2010) A protocol for the investigation of the intracellular Staphylococcus aureus metabolome. Anal. Biochem. 401, 250–259 [DOI] [PubMed] [Google Scholar]
- 20. Liebeke M., Meyer H., Donat S., Ohlsen K., Lalk M.(2010) A metabolomic view of Staphylococcus aureus and its Ser/Thr kinase and phosphatase deletion mutants: involvement in cell wall biosynthesis. Chem. Biol. 17, 820–830 [DOI] [PubMed] [Google Scholar]
- 21. Liebeke M., Pöther D. C., van Duy N., Albrecht D., Becher D., Hochgräfe F., Lalk M., Hecker M., Antelmann H. (2008) Depletion of thiol-containing proteins in response to quinones in Bacillus subtilis. Mol. Microbiol. 69, 1513–1529 [DOI] [PubMed] [Google Scholar]
- 22. Liebeke M., Dörries K., Zühlke D., Bernhardt J., Fuchs S., Pané-Farré J., Engelmann S., Volker U., Bode R., Dandekar T., Lindequist U., Hecker M., Lalk M. (2011) A metabolomics and proteomics study of the adaptation of Staphylococcus aureus to glucose starvation. Mol. BioSyst. 7, 1241–1253 [DOI] [PubMed] [Google Scholar]
- 23. Liebeke M., Brözel V. S., Hecker M., Lalk M. (2009) Chemical characterization of soil extract as growth media for the ecophysiological study of bacteria. Appl. Microbiol. Biotechnol. 83, 161–173 [DOI] [PubMed] [Google Scholar]
- 24. Boisvert F.-M., Ahmad Y., Gierlinski M., Charrière F., Lamont D., Scott M., Barton G., Lamond A. I. (2012) A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell. Proteomics 10.1074/mcp.M111.011429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahler H. C., Friess W., Grauschopf U., Kiese S. (2009) Protein aggregation: pathways, induction factors and analysis. J. Pharm. Sci. 98, 2909–2934 [DOI] [PubMed] [Google Scholar]
- 26. Bernhardt J., Funke S., Hecker M., Siebourg J. (2009) Visualizing Gene Expression Data via Voronoi Treemaps, 2009 Sixth International Symposium on Voronoi Diagrams 233–241 [Google Scholar]
- 27. Michel A., Agerer F., Hauck C. R., Herrmann M., Ullrich J., Hacker J., Ohlsen K. (2006) Global regulatory impact of ClpP protease of Staphylococcus aureus on regulons involved in virulence, oxidative stress response, autolysis, and DNA repair. J. Bacteriol. 188, 5783–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donegan N. P., Thompson E. T., Fu Z., Cheung A. L. (2010) Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 192, 1416–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frees D., Andersen J. H., Hemmingsen L., Koskenniemi K., Bæk K. T., Muhammed M. K., Gudeta D. D., Nyman T. A., Sukura A., Varmanen P., Savijoki K. (2011) New insights into Staphylococcus aureus stress tolerance and virulence regulation from an analysis of the role of the ClpP protease in the strains Newman, COL, and SA564, J. Proteome Res. [DOI] [PubMed] [Google Scholar]
- 30. Clauditz A., Resch A., Wieland K.-P., Peschel A., Götz F. (2006) Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 74, 4950–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Handke L. D., Shivers R. P., Sonenshein A. L. (2008) Interaction of Bacillus subtilis CodY with GTP. J. Bacteriol. 190, 798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stenz L., Francois P., Whiteson K., Wolz C., Linder P., Schrenzel J. (2011) The CodY pleiotropic repressor controls virulence in gram-positive pathogens. FEMS Immunol. Med. Microbiol. 62, 123–139 [DOI] [PubMed] [Google Scholar]
- 33. Wilkins M. R., Gasteiger E., Sanchez J. C., Bairoch A., Hochstrasser D. F. (1998) Two-dimensional gel electrophoresis for proteome projects: the effects of protein hydrophobicity and copy number. Electrophoresis 19, 1501–1505 [DOI] [PubMed] [Google Scholar]
- 34. Rabilloud T., Chevallet M., Luche S., Lelong C. (2010) Two-dimensional gel electrophoresis in proteomics: Past, present and future. J. Proteomics 73, 2064–2077 [DOI] [PubMed] [Google Scholar]
- 35. Kock H., Gerth U., Hecker M. (2004) MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis. Mol. Microbiol. 51, 1087–1102 [DOI] [PubMed] [Google Scholar]
- 36. Gerth U., Kirstein J., Mostertz J., Waldminghaus T., Miethke M., Kock H., Hecker M. (2004) Fine-tuning in regulation of Clp protein content in Bacillus subtilis. J. Bacteriol. 186, 179–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colzani M., Schütz F., Potts A., Waridel P., Quadroni M. (2008) Relative protein quantification by isobaric SILAC with immonium ion splitting (ISIS). Mol. Cell. Proteomics 7, 927–937 [DOI] [PubMed] [Google Scholar]
- 38. Maisonneuve E., Fraysse L., Moinier D., Dukan S. (2008) Existence of abnormal protein aggregates in healthy Escherichia coli cells. J. Bacteriol. 190, 887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maisonneuve E., Ezraty B., Dukan S. (2008) Protein aggregates: an aging factor involved in cell death. J. Bacteriol. 190, 6070–6075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mogk A., Tomoyasu T., Goloubinoff P., Rüdiger S., Röder D., Langen H., Bukau B. (1999) Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 18, 6934–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tabner B. J., Turnbull S., El-Agnaf O., Allsop D. (2001) Production of reactive oxygen species from aggregating proteins implicated in Alzheimer′s disease, Parkinsons disease and other neurodegenerative diseases. Curr. Top. Med. Chem. 1, 507–517 [DOI] [PubMed] [Google Scholar]
- 42. Malhotra J. D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S. W., Kaufman R. J. (2008) Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U.S.A. 105, 18525–18530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabner B. J., El-Agnaf O. M. A., Turnbull S., German M. J., Paleologou K. E., Hayashi Y., Cooper L. J., Fullwood N. J., Allsop D. (2005) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J. Biol. Chem. 280, 35789–35792 [DOI] [PubMed] [Google Scholar]
- 44. Miller R. A., Britigan B. E. (1997) Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 10, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elsholz A. K. W., Hempel K., Pöther D. C., Becher D., Hecker M., Gerth U. (2011) CtsR inactivation during thiol-specific stress in low GC, Gram+ bacteria. Mol. Microbiol. 79, 772–785 [DOI] [PubMed] [Google Scholar]
- 46. Szabo A., Langer T., Schröder H., Flanagan J., Bukau B., Hartl F. (1994) The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. U.S.A. 91, 10345–10349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Doyle S. M., Hoskins J. R., Wickner S. (2007) Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proc. Natl. Acad. Sci. U.S.A. 104, 11138–11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Majerczyk C. D., Dunman P. M., Luong T. T., Lee C. Y., Sadykov M. R., Somerville G. A., Bodi K., Sonenshein A. L. (2010) Direct targets of CodY in Staphylococcus aureus. J. Bacteriol. 192, 2861–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kock H., Gerth U., Hecker M. (2004) The ClpP peptidase is the major determinant of bulk protein turnover in Bacillus subtilis. J. Bacteriol. 186, 5856–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Frees D., Savijoki K., Varmanen P., Ingmer H. (2007) Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63, 1285–1295 [DOI] [PubMed] [Google Scholar]
- 51. Reeves A., Gerth U., Völker U., Haldenwang W. G. (2007) ClpP modulates the activity of the Bacillus subtilis stress response transcription factor, sigmaB. J. Bacteriol. 189, 6168–6175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang C., Li M., Dong D., Wang J., Ren J., Otto M., Gao Q. (2007) Role of ClpP in biofilm formation and virulence of Staphylococcus epidermidis. Microbes Infect. 9, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 53. Frees D., Sørensen K., Ingmer H. (2005) Global virulence regulation in Staphylococcus aureus: pinpointing the roles of ClpP and ClpX in the sar/agr regulatory network. Infect. Immun. 73, 8100–8108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Böttcher T., Sieber S. A. (2009) Structurally refined beta-lactones as potent inhibitors of devastating bacterial virulence factors. Chembiochem 10, 663–666 [DOI] [PubMed] [Google Scholar]
- 55. Kirstein J., Hoffmann A., Lilie H., Schmidt R., Rübsamen-Waigmann H., Brötz-Oesterhelt H., Mogk A., Turgay K. (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 1, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Didier J. P., Cozzone A. J., Duclos B. (2010) Phosphorylation of the virulence regulator SarA modulates its ability to bind DNA in Staphylococcus aureus. FEMS Microbiol. Lett. 306, 30–36 [DOI] [PubMed] [Google Scholar]
- 57. Bronner S., Monteil H., Prévost G. (2004) Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol. Rev. 28, 183–200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.