

Summary
We identified the TMEM132D, SPTA1 and VPS13B genes as frequently mutated and expressed in any progression stage of SCLC, in addition to known tumor suppressor genes, TP53, RB1 and PTEN. These genes could be involved in SCLC development, and their mutated gene products will be promising therapeutic targets in SCLC patients.
Abstract
Small cell lung cancer (SCLC) is the most aggressive type of lung cancer. Only 15% of SCLC patients survive beyond 2 years after diagnosis. Therefore, for the improvement of patients’ outcome in this disease, it is necessary to identify genetic alterations applicable as therapeutic targets in SCLC cells. The purpose of this study is the identification of genes frequently mutated and expressed in SCLCs that will be targetable for therapy of SCLC patients. Exome sequencing was performed in 28 primary tumors and 16 metastatic tumors from 38 patients with SCLCs. Expression of mutant alleles was verified in 19 cases by RNA sequencing. TP53, RB1 and PTEN were identified as being significantly mutated genes. Additional 36 genes were identified as being frequently (≥10%) mutated in SCLCs by combining the results of this study and two recent studies. Mutated alleles were expressed in 8 of the 36 genes, TMEM132D, SPTA1, VPS13B, CSMD2, ANK2, ASTN1, ASPM and FBN3. In particular, the TMEM132D, SPTA1 and VPS13B genes were commonly mutated in both early and late stage tumors, primary tumors and metastases, and tumors before and after chemotherapy, as in the case of the TP53 and RB1 genes. Therefore, in addition to TP53, RB1 and PTEN, TMEM132D, SPTA1 and VPS13B could be also involved in SCLC development, with the products from their mutated alleles being potential therapeutic targets in SCLC patients.
Introduction
Small cell lung cancer (SCLC) is the most aggressive type of lung cancer with extremely poor prognosis (1–3). Therefore, it is critical to develop novel therapies for the improvement of prognosis. Up to the present, extensive genome-wide molecular analyses have been performed in SCLC (4–8). However, targetable genetic alterations have not been defined. This could be due to several reasons. First, mutation rates in SCLCs are very high. Second, except for the frequent inactivation of the TP53 and RB1 genes, the spectra of genes with high mutation frequencies are considerably different among studies. Third, only a small number of clinical SCLC cases have been analyzed to date, due to the small proportion of surgical cases. Fourth, no known druggable mutations have been identified to date. Therefore, we still do not have enough information to select genes with significant clinical importance and with druggable mutations in SCLC.
We previously performed the whole-genome copy number analysis and RNA sequencing of 58 and 42 SCLCs, respectively, and revealed the importance of amplification of MYC family genes and several other genes (8). In this study, we attempted to obtain further information on genes frequently mutated in SCLC through the analysis of 38 SCLC cases. The study confirmed high mutation rates in SCLC, and identified TP53, RB1 and PTEN as significantly mutated genes in SCLCs. Furthermore, by a comparative study of our results with those of two previous studies (6,7), additional 36 genes were identified as being frequently (≥10%) mutated in SCLCs. Therefore, expression of mutated alleles in these genes was validated by whole transcriptome sequencing, and their mutation statuses were investigated in early and late stages of SCLCs, in primary tumors and metastases, and in tumors before and after chemotherapy. Mutated alleles were expressed only in 8 of the 36 genes, and sets of genes mutated in tumors were drastically different between early and late stages SCLCs, between primary tumors and metastases, and also between tumors before and after chemotherapy. We discussed the possible roles of significantly and frequently mutated genes in SCLC development, and defined several therapeutically targetable genes in SCLCs.
Patients and methods
Tissues
Forty-four tumors and the corresponding non-cancerous tissues were obtained from 38 patients with SCLC at surgery or autopsy from 1985 to 2010 at the National Cancer Center Hospital, Tokyo, Japan; Saitama Medical University, Saitama, Japan; University of Tsukuba, Ibaraki, Japan; and hospitals in the Metropolitan Baltimore area in the USA, and kept frozen until DNA and RNA extraction. Clinicopathological characteristics and sample information are summarized in Table 1 and Supplementary Table 1 is available at Carcinogenesis Online, respectively. The tumors were histologically diagnosed according to the 2004 WHO classification and pathologically staged according to the tumor-node metastasis classification of malignant tumors (9,10). Immunohistochemical staining data for neuroendocrine markers, synaptophysin, chromogranine and neural cell adhesion molecule (NCAM), were available in most cases; thus, were added in Supplementary Table 1, available at Carcinogenesis Online. Primary tumors were obtained from 25 cases, and metastases were obtained from 9 cases. Both primary tumors and metastases were obtained in cases 10 and 22. Two metastases were obtained in case 20, while the primary tumors and metastases to three different organs were obtained in case 21. Thirty cases were not treated by chemotherapy/radiotherapy, while the other 7 cases were treated before sampling. In case 10, the primary tumor was obtained before chemotherapy, and the metastasis was obtained after chemotherapy. Genomic DNA was extracted from frozen samples with a QIAamp DNA mini kit (Qiagen, Hilden, Germany). Total RNA was also extracted from frozen samples using TRIzol reagent (Invitrogen, Carlsbad, CA), purified by an RNeasy kit (Qiagen), and reverse-transcribed to cDNA by using the SuperScript III First-Strand Synthesis System (Invitrogen) with random hexamers. This study was performed under the approval of the Institutional Review Board of the National Cancer Center, Tokyo, Japan.
Table 1.
Clinicopathological characteristics of 38 SCLC cases
| Gender | |
| Male/female | 28/10 |
| Age | |
| Median (range) | 67 (56–89) |
| Smoking status | |
| Ever/never | 33/2 (3 unknown) |
| Brinkman index | |
| Median (range) | 1000 (0–2040) |
| Pathological stage | |
| I/II/III/IV | 10/8/10/10 |
| Sampling at | |
| Surgery/autopsy | 36/2 |
| Treatment before sampling | |
| −/+ | 31/8a |
aIn one case, tumors were obtained before and after chemotherapy
Exome sequencing and Data processing
Methods of exome sequencing and data processing are summarized in Supplementary Figure 1 is available at Carcinogenesis Online. One microgram of genomic DNA was fragmented using the Covaris S220. After ligation of the paired end adapter including index, a fraction of 300–350bp was gel-purified and amplified with PCR using the TruSeq DNA Sample Prep Kit (Illumina). Exome capture was performed using the TruSeq Exome Enrichment Kit (Illumina) according to the manufacturer’s protocol. The resulting libraries were subjected to the paired end sequencing of 100-bp reads on the Genome Analyzer IIx (GAIIx) or HiSeq 2000 (Illumina). Mutation calling was performed using the EBCall algorithm (13). Germline variations represented in dbSNP Build 131 or 1000 Genome project (November 2010) and synonymous variations were filtered out. Variants present in the tumor with P < 0.01 by Fisher’s exact test as compared to those in the matched normal tissue were predicted to be somatic mutations. Mean of target coverage in 44 tumors and 38 normal tissues was 97.3 (31.4–170.1) (Supplementary Figure 2, available at Carcinogenesis Online). MutSigCV analysis (14) was performed using expression profile data of 19 tumors analyzed by exome sequencing.
RNA sequencing and data processing
RNA sequencing was performed as described previously (8,15). Expression of mutated alleles detected by exome sequencing of the corresponding tumors was validated by a Bayesian inference method using the samtools and bcftools software (16,17).
Microarray experiments and data processing
Two micrograms of total RNA were labeled using a 5X MEGAscript T7 Kit (Ambion, Austin, TX) and analyzed by U133Plus2.0 arrays (Affymetrix). Data was processed by the MAS5 algorithm as described previously (18).
Results
Significantly mutated genes
Exome sequencing of 44 tumors from 38 SCLC patients identified 9279 protein-altering somatic mutations. In four cases with multiple tumors analyzed, common and unique mutations were detected between primary tumors and metastases and also between metastases of different organs. Therefore, total numbers of different somatic mutations among the multiple tumors in the same patients were considered as the number of somatic mutations in each case. Thirty-eight cases had an average of 244.2 protein-altering somatic mutations (19–1023) with the mean rate of 7.4 mutations (0.6–30.8) per megabase in 5669 genes (Supplementary Figure 3, available at Carcinogenesis Online). Genes with protein altering mutations are listed with their nucleotide sequence changes in Supplementary Table 2, available at Carcinogenesis Online. G/C>T/A transversions occurred most frequently (Supplementary Figure 4, available at Carcinogenesis Online), consistent with the association of tobacco smoking with the occurrence of G/C>T/A transversions. Several software programs are now available to distinguish driver mutations from passenger mutations. We applied the MutSigCV method (14) to identify significantly mutated genes among the 5669 genes by integrating the expression profile data. Then, the TP53, RB1 and PTEN genes were identified as being significantly mutated in SCLCs. TP53 was mutated in 30 cases (78.9%), RB1 in 28 cases (73.7%), and PTEN in five cases (13.2%) (Supplementary Table 3, available at Carcinogenesis Online). Types of TP53, RB1 and PTEN mutations are summarized in Supplementary Table 4, available at Carcinogenesis Online. Previously, Peifer et al. (6). identified 22 significantly mutated genes, and Rudin et al. (7). identified another set of 22 significantly mutated genes in SCLCs. Although the criteria of ‘significantly mutated gene’ are different among three studies, both TP53 and RB1 were identified in all studies, while PTEN was identified only in this study (Supplementary Table 5, available at Carcinogenesis Online). PTEN was mutated in 2/27 (7.4%) and 2/30 (6.7%) in those studies, respectively (Supplementary Table 3, available at Carcinogenesis Online). Therefore, although PTEN mutations could be significant, its mutation frequency is much lower than those of TP53 and RB1 in SCLCs.
Frequently mutated genes
A total of 263 genes were mutated in ≥10% of the 38 cases (Figure 1). However, the power to detect frequently mutated genes could be small because of a small sample size in this study and a high background mutation frequency in SCLC. To increase the power of defining genes frequently mutated in SCLC, we further selected frequently mutated genes (≥10%) from two previous studies for whole exome sequencing of SCLC, in which the results of 27 and 30 SCLC cases were reported, respectively (6,7). Similarly high mutation rates of 7.4 and 5.5 per megabase, respectively, were reported in those studies, justifying a combined analysis. A total of 331 genes and 230 genes, respectively, were identified as being mutated in ≥10% of SCLC cases in those studies (Figure 1). Sets of frequently mutated genes were considerably different among three studies possibly due to the difference in analytical methods used and the small number of cases analyzed in each study, and also due to the high mutation rates in SCLCs. SCLC samples were obtained mostly from Japanese in this study, while those were from Europeans/Americans in the previous studies. Therefore, it was also possible that such differences could be due to the ethnic and geographic differences of patients. Similarities in mutation frequencies and diversities in spectra of mutated genes among three studies indicated that genes commonly and frequently mutated in SCLC could be further selected by combining these data. Therefore, we further selected genes mutated in ≥10% of 95 SCLC cases, and also in ≥10% of 38 cases in this study and of 27 and 30 cases in two previous studies, respectively. A total of 38 genes were identified as being frequently mutated (≥10%) in all three studies as well as in a total of 95 cases (Figure 1 and Supplementary Table 3, available at Carcinogenesis Online). There was a low variability in the mutation frequencies of the 38 genes among three studies, and TP53 and RB1 were the first and second most frequently mutated genes in all studies. Thus, we concluded that, in addition to TP53 and RB1, there are 36 genes that are frequently mutated in SCLCs irrespective of the geographic and ethnic differences of patients. PTEN was not included in the 38 genes, because of low frequencies of mutations (<10%) in two other studies.
Figure 1.

Numbers of genes frequently mutated in SCLC: Comparison between this study and two previous studies (a, ref 6; b, ref 7). Genes with mutation frequencies ≥10% of cases were selected in each study. Then, similarities and differences of genes selected were compared among three studies.
Mutated genes with expression
MutSigCV analysis identified only three genes as being significantly mutated in SCLC. However, it is possible that there are additional genes whose mutations are also involved in SCLC development among the 36 genes frequently mutated in SCLCs. Indeed, four of the 36 genes, TMEM132D, HCN1, SPHKAP and COL11A1, were identified as being significantly mutated in SCLCs by Peifer et al. (6), and 2 of the 36 genes, COL22A1 and TMEM132D, were also identified by Rudin et al. (7). Expression of the mutated allele is an important factor to support the significance of mutations in cancer development and to consider target therapies against mutated gene products. We previously performed whole transcriptome sequencing in 19 cases analyzed in this study (8). Therefore, transcripts from mutated alleles were searched for in the sequencing data of 36 frequently mutated genes in addition to TP53, RB1 and PTEN (Figure 2). Expression of mutated alleles in the TP53 and RB1 genes was validated in several SCLC cases. PTEN was mutated only in one case, and expression of the mutated allele was not detected in this tumor. In addition, mutated alleles were expressed in 8 of the 36 genes, TMEM132D, SPTA1, VPS13B, CSMD2, ANK2, ASTN1, ASPM and FBN3, in at least one of the 19 tumors (Figure 2).
Figure 2.
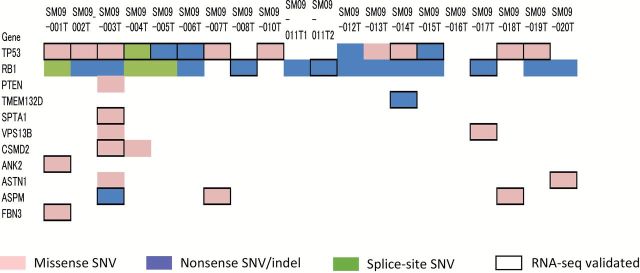
Expression of mutated alleles in significantly or frequently mutated genes in SCLC. Twenty tumors from 19 patients were analyzed by both exome sequencing and RNA sequencing. Types of mutations detected in tumors by exome sequencing are indicated by colored boxes, and expression of mutated alleles validated by RNA sequencing (RNA-seq validated) is indicated by black open squares.
Mutated genes in stage I SCLCs before chemotherapy/radiotherapy
Driver mutations essential for SCLC development and its maintenance should be detected in early stage tumors before chemotherapy/radiotherapy. Therefore, we next analyzed the timing of mutation occurrence by considering the pathological stages and the effect of chemotherapy/radiotherapy. Average numbers of mutated genes tended to increase with stage progression (Supplementary Figure 5, available at Carcinogenesis Online), and significant difference in the number of mutated genes was observed between stage I tumors and stages II–IV tumors (P = 0.048 by the Student’s t-test). The mean number of mutated genes in stage I tumors was 177.7, while that in stages II–IV tumors was 245.9 (Figure 3A). The result was consistent with the concept of sequential accumulation of somatic mutations during tumor progression (19–21). A total of 727 genes were mutated commonly in both stages I and II–IV tumors, while other 821 genes were mutated only in stage I tumors. It was noted that 4121 (72.7%) of the 5669 genes identified in this study were mutated only in stages II–IV tumors but not in stage I tumors. This result strongly indicates that most of mutations detected in SCLC cells have occurred during tumor progression, thus, may not be necessary for tumor development, although the small number of early stage tumors may influence this assumption. TP53 and RB1 were mutated in any progression stages of tumors, supporting their importance in tumor development and its maintenance. On the other hand, PTEN mutations were detected only in stages II–IV tumors. Among eight frequently mutated and expressed genes, four genes, TMEM132D, SPTA1, VPS13B, ASPM, were mutated in both stages I and II–IV tumors, while the remaining four genes, CSMD2, ANK2, ASTN1, FBN3, were mutated only in stages II–IV tumors but not in stage I tumors. Therefore, although the frequency of their mutations were not significantly different between stage I tumors and stage II–IV tumors, the TP53, RB1, TMEM132D, SPTA1, VPS13B and ASPM genes could play more important roles in SCLC development than other genes.
Figure 3.
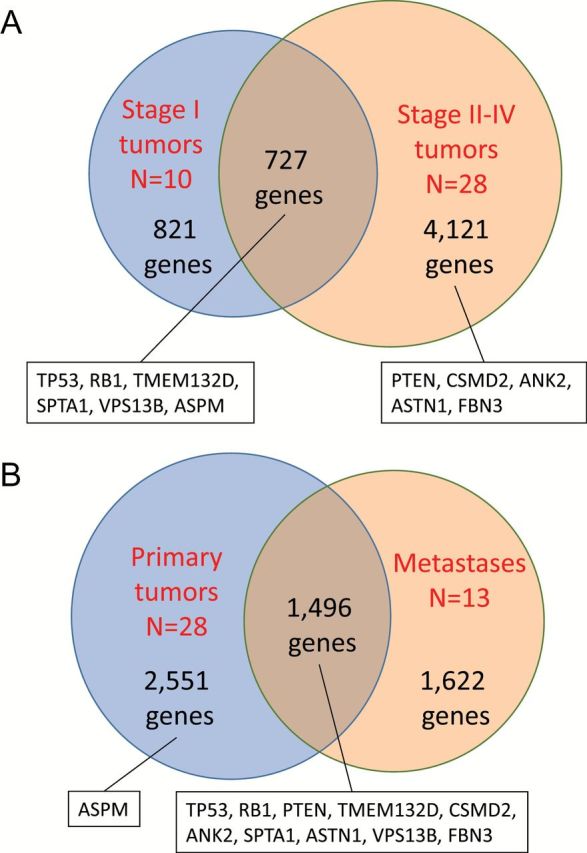
Number of genes mutated in early and late stages of SCLCs. (A) Comparison of mutated genes between stage I tumors and stage II–IV tumors. (B) Comparison of mutated genes between primary tumors and metastases. Mutation status of three significantly mutated genes and eight frequently mutated and expressed genes are indicated in the boxes.
We further investigated whether the 5669 genes were mutated before or after chemotherapy/radiotherapy (Supplementary Figure 6, available at Carcinogenesis Online). In case 10, mutation data in two tumors obtained before and after chemotherapy were analyzed separately. Importantly, 722 genes were mutated only in tumors after chemotherapy/radiotherapy, and 38 of them showed significantly higher mutation frequencies in tumors after therapy than in tumors before therapy (data not shown). Therefore, it is possible that these mutations have occurred after therapy. However, none of the eight genes frequently mutated and expressed in SCLCs and the TP53, RB1 and PTEN genes showed higher mutation frequencies in tumors after therapy than in tumors before therapy. In particular, mutations in the VPS13B and ASPM genes were detected only in tumors before therapy and not in tumors after therapy. Thus, none of the 11 genes was considered to be secondary mutated after or during chemotherapy/radiotherapy.
Genes mutated in both primary tumors and metastases
Driver mutations essential for SCLC development and its maintenance should be detected in both primary tumors and metastases. Both primary tumors and metastases were analyzed in three cases; therefore, mutation data in primary tumors and metastases in these cases were analyzed separately. The mean number of mutated genes in 28 primary tumors was 194.1, while that in 13 metastases was 298.0. Although the difference was not statistically significant (P = 0.073 by the Student’s t-test), metastases tended to carry more mutations than primary tumors (Figure 3B). This result was also consistent with the concept of sequential accumulation of somatic mutations during tumor progression (19–21). Although 1622 of the 5669 genes were mutated only in the metastases, 7 genes frequently mutated and expressed in SCLCs and the TP53, RB1 and PTEN genes were commonly mutated in both primary tumors and metastases. In contrast, ASPN mutations were detected only in primary tumors but not in metastases suggesting their possibility of being passengers.
Genetic heterogeneity among multiple tumors in the same patients
We next compared the accumulated genetic alterations among multiple tumors in four cases. The results strongly support the concept of clonal and parallel evolution of primary tumors and metastases (19–21). Namely, common and unique mutations were detected between primary tumors and metastases, and also between metastases of different organs (Figure 4; Supplementary Figure 7, available at Carcinogenesis Online). In particular, in the analysis of the primary tumor and three different metastases from a single patient, drastic heterogeneity in the accumulated mutations was observed among the four tumors (Figure 4). The TP53, TMED132D and SPTA1 genes, which were defined as being frequently mutated and expressed in SCLCs, were mutated in all the tumors. In contrast, a mutation of another gene frequently mutated and expressed in SCLCs, FBN3, was detected only in a hilar lymph node metastasis but not in the primary tumor, a liver metastasis, and a para-aortic lymph node metastasis. Therefore, FBN3 mutation could have occurred during tumor progression after TP53, TMED132D and SPTA1 mutations.
Figure 4.
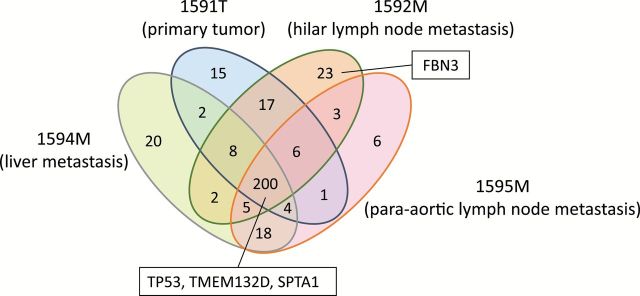
Heterogeneity in the accumulated mutations among multiple tumors in a single SCLC patient. The primary tumor (1591T), a hilar lymph node metastasis (1592M), a liver metastasis (1594M), and a para-aortic lymph node metastasis (1595M) from a single patient were analyzed for accumulated mutations by exome sequencing. Numbers in each region indicate the numbers of genes with mutations detected in tumors. Mutation status of one of significantly mutated gene, TP53, and three of frequently mutated and expressed genes, TMEM132D, SPTA1 and FBN3, are indicated in the boxes.
Discussion
MutSigCV analysis revealed that TP53, RB1 and PTEN were significantly mutated genes in SCLC. These genes are well-known tumor suppressors involved in the development of various cancers, including SCLC. Therefore, the present results further support that these genes are involved in SCLC development. However, this analysis failed to identify various other genes previously identified as being significantly mutated in SCLCs (6,7). Therefore, we further selected genes frequently mutated and expressed in SCLCs and investigated their clinical significance by analyzing their mutation status in various stages of SCLCs. As in the case of TP53 and RB1, the TMEM132D, SPTA1 and VPS13B genes were mutated in any stage of tumor progression and their mutated alleles were expressed in tumors. Recent comparative genomic analyses of primary and metastatic non-SCLCs and colorectal cancers revealed high concordance rates of driver alterations and low concordance rates of likely passenger alterations (22,23). Therefore, in addition to TP53, RB1 and PTEN, TMEM132D, SPTA1 and VPS13B could be also mutated as drivers in SCLC development. In particular, TMED132D was considered as being a significantly mutated gene in the two previous studies (6,7). Since mutated products from the TMEM132D, SPTA1 and VPS13B genes are often expressed in SCLCs, those products could be novel therapeutic targets in SCLC patients.
In this study, TP53 mutations were not detected in 8 of the 38 cases (21.2%), and RB1 mutations were not detected in 10 of the 38 cases (26.3%). The results indicate the presence of a noble subset of SCLCs without TP53 mutations and/or RB1 mutations. Therefore, it is very important to clarify whether the TP53 and RB1 genes are inactivated or not inactivated in tumors without detectable mutations by exome sequencing. We previously performed SNP array analysis in 19 of the 38 SCLC cases analyzed in this study (8). Therefore, the presence/absence of loss of heterozygosity (LOH) at the TP53 and RB1 loci were examined in tumors without TP53 and/or RB1 mutations (Supplementary Table 6, available at Carcinogenesis Online). LOH of the TP53 locus was detected in 4 of 5 cases without TP53 mutations, and LOH of the RB1 locus was detected in 3 of 4 cases without RB1 mutations. Therefore, it is likely that undetectable TP53 and RB1 mutations are present in the cases without their mutations. However, it is also possible that there is still a noble small subset of SCLCs without mutations of the TP53 and/or RB1 genes. Further molecular analyses, such as whole-genome sequencing and methylation array analysis in combination with mRNA expression array analysis, will be able to reveal the presence/absence of a noble subset of SCLCs.
Similar studies were also performed to predict whether the TMEM132D, SPTA1 and VPS13B genes function as oncogenes or tumor suppressor genes. LOH, loss of wild-type allele as well as nonsense mutation were found in the TMEM132D gene, while missense mutations without allelic losses or with chromosomal gain were common in the SPTA1 and VPS13B genes. Therefore, the TMEM132D gene is likely to function as a tumor suppressor gene, whereas the SPTA1 and VPS13B genes are likely to function as oncogenes. However, since involvement of the TMEM132D, SPTA1 and VPS13B genes in cancer development is unknown at present, functional studies will be necessary to elucidate the pathogenic significance of their mutations in SCLC development. The TMEM132D (transmembrane protein 132D) gene encodes a single-pass transmembrane protein, the SPTA1 (spectrin, alpha, erythrocytic 1) gene encodes an actin crosslinking and molecular scaffold protein that links the plasma membrane to the actin cytoskeleton, spectrin, and the VPS13B (vascular protein sorting 13 homolog B) gene also encodes a potential transmembrane protein (NCBI databases: http://www.ncbi.nlm.nih.gov). The results indicate that mutated proteins are expressed on the membrane of SCLC cells. Therefore, although functional roles of these mutated gene products are presently unknown, the products could be appropriate targets of immunotherapy in SCLC patients.
Our study has several limitations. The number of cases analyzed in this study was small; therefore, we mainly analyzed the status of genes mutated in ≥10% of SCLCs. For the identification of targetable mutations in SCLCs, we should also analyze the status of genes with low frequency of mutations in SCLCs. As reported previously (6,7), we also found mutations in the CREBBP (2 cases), EP300 (2 cases), Ephrin family (19 cases in 10 genes), and FLT family (7 cases in 3 genes) genes with low frequencies (<10%). Therefore, it is absolutely necessary to analyze a larger number of SCLCs to define the prevalence and clinical significance of their mutations. However, it is difficult to collect SCLC samples for molecular analysis, these results will be highly informative to further define genes targetable for therapy of SCLC patients.
Supplementary material
Supplementary Table 1–6 and Figures 1–7 can be found at http://carcin.oxfordjournals.org/
Funding
Ministry of Health, Labor, and Welfare, Japan, for Practical Research for Innovative Cancer Control (H26-practical-general-007/094); ISIS (IIS13/00849) and FIS (PI13/00849) grants (Institute of Health Carlos III) in Spain, co-financed by the European Regional Development Funds (FEDER) of the European Union.
Supplementary Material
Acknowledgements
R.I. is an awardee of Research Resident Fellowship from the Foundation for Promotion of Cancer Research for the third-term comprehensive 10-year strategy for Cancer Control, Japan.
Conception and design: R.I., T.K., J.Y. Collection and assembly of data: R.I., Y.T., T.S., K.T., S.M., K.T., Y.N., R.N., M.N., C.C.H. and A.I.R. Data analysis and interpretation: R.I., R.Y., S.I., S.M., H.T. and T.Y. Manuscript writing: all authors. Final approval of manuscript: all authors. Administrative support: T.Y.
Conflict of Interest Statement: Employment, leadership, or stock ownership: none; Honoraria: R.N., EISAI; R.Y. and S.I.; Fujifilum; Consultant or advisory role: R.N., Roche; Speaker’s bureau: R.N., Chugai; Research funding, or patents, royalties, other intellectual property: none.
Glossary
Abbreviations
- LOH
loss of heterozygosity
- SCLC
small cell lung cancer
References
- 1. Jemal A., et al. (2011) Global cancer statistics. CA. Cancer J. Clin., 61, 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Kalemkerian GP, et al. (2013) Small cell lung cancer. J Natl Compr Canc Netw., 11, 78–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Meerbeeck J.P., et al. (2011) Small-cell lung cancer. Lancet, 378, 1741–1755. [DOI] [PubMed] [Google Scholar]
- 4. Pleasance E.D., et al. (2010) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature, 463, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sos M.L., et al. (2012) A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc. Natl. Acad. Sci. USA, 109, 17034–17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peifer M., et al. (2012) Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet., 44, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rudin C.M., et al. (2012) Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet., 44, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iwakawa R., et al. (2013) Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer, 52, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sobin LH, et al. (2009) International Union Against Cancer: TNM Classification of Malignant Tumours. 7th edn Wiley-Blackwell, West Sussex, UK. [Google Scholar]
- 10. Travis WD, et al. (2004) World Health Organization Classification of Tumors: Pathology and Genetics; Tumours of the Lung, Pleura, Thymus and Heart. IARC Press, Lyon, France. [DOI] [PubMed] [Google Scholar]
- 11. Phelps R.M., et al. (1996) NCI-Navy Medical Oncology Branch cell line data base. J. Cell. Biochem., (suppl), 24, 32–91. [DOI] [PubMed] [Google Scholar]
- 12. Iwakawa R., et al. (2011) MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clin. Cancer Res., 17, 1481–1489. [DOI] [PubMed] [Google Scholar]
- 13. Shiraishi Y., et al. (2013) An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res., 41, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawrence M.S., et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kohno T., et al. (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat. Med., 18, 375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H, et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 16, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H. (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okayama H., et al. (2012) Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res., 72, 100–111. [DOI] [PubMed] [Google Scholar]
- 19. Burrell R.A., et al. (2013) The causes and consequences of genetic heterogeneity in cancer evolution. Nature, 501, 338–345. [DOI] [PubMed] [Google Scholar]
- 20. Yokota J. (2000) Tumor progression and metastasis. Carcinogenesis, 21, 497–503. [DOI] [PubMed] [Google Scholar]
- 21. Takahashi K., et al. (2007) Clonal and parallel evolution of primary lung cancers and their metastases revealed by molecular dissection of cancer cells. Clin. Cancer Res., 13, 111–120. [DOI] [PubMed] [Google Scholar]
- 22. Vignot S., et al. (2013) Next-generation sequencing reveals high concordance of recurrent somatic alterations between primary tumor and metastases from patients with non-small-cell lung cancer. J. Clin. Oncol., 31, 2167–2172. [DOI] [PubMed] [Google Scholar]
- 23. Vakiani E., et al. (2012) Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J. Clin. Oncol., 30, 2956–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.