

ABSTRACT
Neurospora crassa cpc-1 and Saccharomyces cerevisiae GCN4 are homologs specifying transcription activators that drive the transcriptional response to amino acid limitation. The cpc-1 mRNA contains two upstream open reading frames (uORFs) in its >700-nucleotide (nt) 5′ leader, and its expression is controlled at the level of translation in response to amino acid starvation. We used N. crassa cell extracts and obtained data indicating that cpc-1 uORF1 and uORF2 are functionally analogous to GCN4 uORF1 and uORF4, respectively, in controlling translation. We also found that the 5′ region upstream of the main coding sequence of the cpc-1 mRNA extends for more than 700 nucleotides without any in-frame stop codon. For 100 cpc-1 homologs from Pezizomycotina and from selected Basidiomycota, 5′ conserved extensions of the CPC1 reading frame are also observed. Multiple non-AUG near-cognate codons (NCCs) in the CPC1 reading frame upstream of uORF2, some deeply conserved, could potentially initiate translation. At least four NCCs initiated translation in vitro. In vivo data were consistent with initiation at NCCs to produce N-terminally extended N. crassa CPC1 isoforms. The pivotal role played by CPC1, combined with its translational regulation by uORFs and NCC utilization, underscores the emerging significance of noncanonical initiation events in controlling gene expression.
KEYWORDS: Neurospora, filamentous fungi, gene regulation, molecular genetics, translational control
IMPORTANCE
There is a deepening and widening appreciation of the diverse roles of translation in controlling gene expression. A central fungal transcription factor, the best-studied example of which is Saccharomyces cerevisiae GCN4, is crucial for the response to amino acid limitation. Two upstream open reading frames (uORFs) in the GCN4 mRNA are critical for controlling GCN4 synthesis. We observed that two uORFs in the corresponding Neurospora crassa cpc-1 mRNA appear functionally analogous to the GCN4 uORFs. We also discovered that, surprisingly, unlike GCN4, the CPC1 coding sequence extends far upstream from the presumed AUG start codon with no other in-frame AUG codons. Similar extensions were seen in homologs from many filamentous fungi. We observed that multiple non-AUG near-cognate codons (NCCs) in this extended reading frame, some conserved, initiated translation to produce longer forms of CPC1, underscoring the significance of noncanonical initiation in controlling gene expression.
INTRODUCTION
General amino acid control (GAAC) in fungi activates amino acid biosynthetic gene expression in response to amino acid limitation (1, 2). This regulatory pathway was originally called cross-pathway control in Neurospora crassa and general control in Saccharomyces cerevisiae (3). N. crassa cpc-1 and S. cerevisiae GCN4 specify homologous bZIP transcription factors that were identified using forward genetics based on their function to transcriptionally activate amino acid biosynthetic genes in response to amino acid limitation or imbalance.
Both N. crassa CPC1 and yeast Gcn4p contain a transcription activation domain, a basic DNA binding domain, and a leucine zipper region involved in dimerization. Genes regulated by CPC1 or GCN4 contain the general control response element (GCRE) sequence TGA(C/G)TCA or a similar sequence (3, 4). A comparative study of S. cerevisiae Gcn4p, Candida albicans Gcn4p, and N. crassa CPC1 revealed that many genes were regulated by these factors in each organism and that the common core of regulated genes was mostly amino acid biosynthetic genes (5). N. crassa cpc-1, like Aspergillus nidulans cpcA and C. albicans GCN4 but unlike S. cerevisiae GCN4, appears transcriptionally autoregulated in response to amino acid limitation (5–8), and these fungal cpc-1 genes contain GCRE sequences in their 5′ regions implicated in transcriptional autoregulation.
The translational control of GCN4 in response to amino acid limitation is the canonical example of how upstream open reading frames (uORFs) mediate regulation of translation via control of reinitiation (1, 9, 10). Four uORFs affect the progression of ribosomes through the 5′ leader of GCN4 mRNA to regulate GCN4 expression in response to amino acid limitation. uORF1 acts as a positive regulatory element to facilitate reinitiation, while uORF4 strongly inhibits the translation of GCN4. uORF2 and uORF3 play relatively minor roles. In vivo experiments (11) and cell-free translation assays (12) confirm that translation of uORF1 generates reinitiating ribosomes that can start translation at either uORF4 or GCN4 and that translation of uORF4 is incompatible with reinitiation at the GCN4 start codon. The phosphorylation of initiation factor eIF2α (α subunit of eukaryotic initiation factor 2) by the GCN2 kinase in response to amino acid limitation causes ribosomes to scan past uORF4 and to increase reinitiation at the GCN4 start codon. Fungal homologs of GCN4 contain at least two uORFs, and it is generally thought that these perform similar functions as GCN4 uORF1 and uORF4. ATF4, a mammalian homolog of GCN4, also contains two uORFs, and these also function similarly to GCN4 uORF1 and uORF4 (13, 14).
N. crassa cpc-1 expression is known to be translationally controlled in response to histidine limitation as determined by polysome association analyses (15). Also, N. crassa cpc-3, the functional homolog of S. cerevisiae GCN2, is required for the GAAC response, and disruption of cpc-3 abolishes the increase of CPC1 protein in response to amino acid starvation (16). These studies are consistent with translational regulation of cpc-1 through its uORFs occurring similarly to that of S. cerevisiae GCN4.
An additional consideration for regulation of cpc-1 is the discovery that the CPC1 reading frame could be extended at its amino terminus if a near-cognate non-AUG start codon (NCC) was used to initiate translation (17). NCCs are known to be used as initiation codons (18–23), and their significance is actively being explored (24–28). In other organisms, the use of NCCs appears to increase in response to conditions that reduce the stringency of start codon selection (29–32).
Here, we used an N. crassa cell-free translation system to show that N. crassa cpc-1 uORF1 and uORF2 act analogously to uORF1 and uORF4, respectively, in S. cerevisiae GCN4 in that ribosomes reinitiate efficiently after translating uORF1 but not uORF2. We also discovered and identified conserved potential N-terminal extensions in the cpc-1 homologs from a much larger group of fungi, including Pezizomycotina and Basidiomycota, but not yeast. Multiple NCCs, some well conserved and in optimal initiation contexts, which potentially initiate the extension of the N. crassa cpc-1 homolog were examined both in vitro and in vivo. The positions of these NCCs indicate that their utilization could bypass the translational inhibitory effect of uORF2. We observed that four of the identified NCCs were used in vitro and that, as predicted, their use abrogated the inhibitory effect of uORF2. Evidence for NCC utilization in vivo was also obtained. These findings indicate that, in addition to translational control via uORFs, the filamentous fungi possess other translational mechanisms to produce different CPC1 isoforms.
RESULTS
Bioinformatic analyses of fungal cpc-1.
While studying the regulation of cpc-1 by its uORFs in N. crassa extracts, using a construct in which the wild-type (WT) 5′ leader of cpc-1 was fused with the open reading frame of firefly luciferase (LUC), we observed a band of predicted size and a band ~20 kDa larger than predicted (Fig. 1A). This prompted a more careful examination of the mRNA 5′ leader sequence. We found that the CPC1 reading frame extended far upstream (Fig. 1B), without any in-frame stop codons, to the major mapped transcription initiation site, which is located 703 nucleotides (nt) 5′ of the predicted AUG for the main open reading frame (designated mAUG and mORF, respectively). We previously noted that N. crassa cpc-1 could hypothetically use upstream near-cognate start codons for initiation (17). We next compiled partial or complete sequences of cpc-1 homologs from 108 Pezizomycotina species: 100 sequences included the region spanning from uORF1 to a position downstream of the mAUG and were analyzed further. All homologs contain two AUG-initiated uORFs, with uORF1 spanning 3 to 6 codons and uORF2 spanning 35 to 70 codons, including their stop codons. Surprisingly, in all cases, the reading frame for CPC1 could be substantially N-terminally extended without encountering an in-frame stop. The shortest extension of the CPC1 ORF without encountering an in-frame stop codon is 160 codons in Leptosphaeria maculans. We note that some automated annotations of CPC1 homologs include this N-terminal extension (e.g., XP_001906068, EGR46729, and EKJ70155), but annotations do not resolve where initiation occurs. The presence of this feature in both Sordariomycetes and Eurotiomycetes suggests that it was present in their last common ancestor and possibly earlier; the last common ancestor of all Pezizomycotina is estimated to have lived at least 320 million years ago (33).
FIG 1 .

N-terminal extensions of Pezizomycotina CPC1. (A) cpc-1–luc mRNA produces a larger product in vitro than luc mRNA. mRNA templates were used to program N. crassa in vitro translation extracts, and [35S]Met-labeled products were analyzed on a 12% NuPAGE gel. Translation of mRNA for N. crassa cpc-1–luc (in which the 5′ leader of cpc-1 plus the first three codons of its mORF are fused in frame to firefly luciferase) and that for luc were compared to a no-mRNA control. The positions of full-length firefly LUC (arrowhead) and the larger CPC1-LUC product (asterisk) are indicated. (B) Schematic diagram of the N. crassa cpc-1 mRNA. Each reading frame is on a separate line. Frame 1 specifies CPC1 (black rectangle). uORF1and uORF2 (blue rectangles) initiate from uAUG1 and uAUG2, respectively, in other reading frames. AUG codons are indicated by green bars, and stop codons are indicated by red bars. NCCs in frame 1 upstream of uORF2 are indicated by magenta bars and are numbered 1 to 8. The approximate position of the 3′-most stop codon upstream of uORF2 and in frame with the main ORF (present in Cordyceps bassiana) is indicated (dashed red bar). The number of sequences used for comparisons is shown in parentheses. Features are drawn to scale. The nucleotide sequence of the N. crassa cpc-1 5′ leader is given in Fig. S6 in the supplemental material. (C to J) Frequency WebLogo of the conservation of the initiation contexts, from −6 to +4, of all N. crassa genes initiated with AUG (C), all A. nidulans genes initiated with AUG (D), Pezizomycotina cpc-1 uAUG1 (E), Pezizomycotina cpc-1 uAUG2 (F), Pezizomycotina cpc-1 mAUG (G), Pezizomycotina NCC6 (CUG) (H), Pezizomycotina NCC7 (AUU) (I), and Pezizomycotina NCC8 (ACG) (J). Letter heights are proportional to the frequency of occurrence of each nucleotide at each position. The crucial positions −3 and +4 are indicated in red underneath the frequency plots. Data used to calculate consensus AUG initiation context for N. crassa and A. nidulans were obtained from the Transterm database (61). (K) The amino acid sequence encoded by the N. crassa mRNA in the CPC1 reading frame, starting from the 5′ end of the mRNA and ending with the first in-frame stop codon. mAUG indicates the annotated cpc-1 initiation codon; upstream NCC6 (uNCC6), uNCC7, and uNCC8 are also indicated. The approximate positions of uORF1 and uORF2 (which are in other reading frames) are indicated by blue lines. The C-terminal bZIP domain of CPC1 is indicated in red font. The regions analyzed in Fig. S3A and B are bracketed by dashed red lines. The highly conserved patch specifically marked in Fig. S3A is bracketed by a dashed orange line. The level of conservation of each residue from the alignment of homologs from 95 species (those used to construct the tree in Fig. S1A) is shown below the amino acid sequence and was generated using ClustalX2. The conservation, expressed as percent amino acid identity, is indicated on the right side of each alignment.
Based on the mechanism of translational control of S. cerevisiae GCN4, control of cpc-1 would involve ribosomes initiating at uORF1 and reinitiating at uORF2 under amino acid-sufficient conditions. When eIF2α phosphorylation levels increase in response to amino acid limitation, ribosomes would reinitiate at the downstream cpc-1 mAUG instead of uORF2. Remarkably, without exception in the Pezizomycotina, there is no stop codon in the reading frame of the mORF between the uORF2 AUG and the mAUG. The in-frame stop codon closest to uAUG2 (Cordyceps bassiana) is 101 nt upstream of it. Thus, the potential amino-terminal extensions of CPC1 are encoded upstream of uORF2 (Fig. 1B).
Initiation upstream of the uORF2 AUG could produce N-terminally extended isoforms of CPC1 whose synthesis would not be subject to inhibition by uORF2. We searched for potential start codons in this region of N. crassa cpc-1 mRNA that were in frame with the predicted mAUG. Eight NCCs fulfilling these criteria were identified—three AUC (NCC1, NCC3, and NCC4), two ACG (NCC2 and NCC8), two AUU (NCC5 and NCC7), and one CUG codon (NCC6) (Fig. 1B). We next searched for potential NCCs in similar regions of cpc-1 transcripts from all Pezizomycotina (see Fig. S1 in the supplemental material) and compared their conservation levels. Three NCCs in N. crassa showed particularly deep conservation—the closest to the CPC1 AUG, an ACG (NCC8), is perfectly conserved in 98 of 100 species; NCC7, an AUU, is conserved in 74 of 100 species (as AUU in 64 and AUC or AUA in 10); NCC6, a CUG, is conserved in 77 of 100 species (as CUG in 73 and UUG in 4) (Fig. S1 and S2). In no case was there an in-frame stop codon between these three conserved NCCs and the mAUG. The three conserved NCCs also show a clear pattern of fungal branch-specific distribution: the minority of homologs lacking both AUU and CUG NCCs clustered separately from the other homologs (Fig. S1). The other five NCCs showed sporadic conservation and were found only in species that were closely related to N. crassa. None of the N. crassa NCCs appeared conserved in the two most distant Pezizomycotina, Arthrobotrys oligospora and Tuber melanosporum (Fig. S1). However, even these two species’ CPC1 homologs contain multiple NCCs upstream of uORF2 and in frame with the mORF—6 NCCs in A. oligospora and 7 NCCs in T. melanosporum (Fig. S2C).
Superimposing conservation of NCCs in cpc-1 homologs on the phylogenetic trees of Pezizomycotina and Agaricomycetes. (A) Phylogeny of Pezizomycotina based on CPC1 amino acid sequences and conservation of NCC6, NCC7, and NCC8. The unrooted phylogenetic tree is based on the amino acid sequences (from the 5′ end to the mORF stop codon when no in-frame stop codon is present or from the stop codon in the 5′ region to the mORF stop codon) of the cpc-1 homologs from the Pezizomycotina species indicated on the right of each branch. Species names are coded with respect to which NCCs are identical to N. crassa: black, NCC6, NCC7, and NCC8; blue, NCC7 and NCC8; green, NCC6 and NCC8; red, NCC8; orange, no identical NCCs. (B) Phylogeny of Agaricomycetes based on CPC1 amino acid sequences and conservation of a single NCC. The unrooted phylogenetic tree is based on the amino acid sequences (from the 5′ end to the mORF stop codon or from the stop codon in the 5′ region to the mORF stop codon) of the cpc-1 homologs from the Agaricomycetes species indicated on the right of each branch. The asterisk indicates a sequence presumed to derive from an unknown Agaricomycetes species contaminating the Pinus taeda EST library. Species names in black, NCC = AUU; names in blue, NCC = UUG; in orange, NCC = AUA; in red, NCC = ACG or CUG. The trees were generated using ClustalX. Bootstrap values are given for key nodes in Pezizomycotina. The scale represents the divergence rate at each residue. Download FIG S1, PDF file, 0.2 MB (160.5KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Conservation of NCCs in cpc-1 homologs of different fungi. (A and B) Nucleotide sequences used for generating WebLogo in Fig. 1 and S5, respectively. Species identifiers of the sequences used are indicated on the left. An asterisk next to the species name indicates sequence presumed to derive from unknown Pezizomycotina or Agaricomycetes that is contaminating an EST library of the indicated species (e.g., Agaricomycetes sequence contaminating Pinus taeda library). Presumed start codons are highlighted in green. Sequences from species names in quotation marks are presumed contaminants of fungal origin because they cluster with cpc-1 from Pezizomycotina or Agaricomycetes even though the name of the sequenced species is a plant. Alignments of the 10 nucleotides including the AUG or NCC start codons and the nucleotides that define the initiation contexts (−6 to +4) which were used to generate the WebLogo in Fig. 1C to J and S5D and E are shown for cpc-1/GCN4 homologs from uAUG1 uAUG2, mAUG, NCC6, NCC7, and NCC8 from Pezizomycotina (A) and the single NCC from Agaricomycetes (B). (C) Nucleotide sequences of the 5′ regions of cpc-1 homologs from the upstream in-frame stop codon to the uAUG in Tuber melanosporum and Arthrobotrys oligospora. NCCs are highlighted in green. Download FIG S2, PDF file, 0.3 MB (265KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We next examined the conservation of the initiation contexts for the three conserved NCCs and for the uORF1 AUG (uAUG1), uORF2 AUG (uAUG2), and mAUG. The preferred initiation context in N. crassa (Fig. 1C), which is considered optimal, is similar to the preferred context in the relatively distant Aspergillus fumigatus (34) and Aspergillus nidulans (as shown in Fig. 1D). uAUG1 and uAUG2 are in conserved optimal contexts (Fig. 1E and F and S2A), consistent with their presumed roles in regulating CPC1 translation through controlling reinitiation. Conservation of mAUG context is weaker, but the consensus is still near optimal (Fig. 1G). Of the three conserved NCCs, NCC8, which is closest to the mAUG and is the most conserved, showed the highest context conservation (Fig. 1J and S2A). The consensus initiation context of NCC8 in species that we examined is nearly optimal (nucleotides −4, −3, −1, and +4 match the consensus). The most important nucleotides, A at position −3 and G at position +4, are perfectly conserved in all Pezizomycotina that have NCC8. Lower context conservation is observed for NCC7 and NCC6 (Fig. 1H and I), although their consensus initiation contexts remain nearly optimal.
One question raised by the potential N-terminal extensions of cpc-1 homologs is whether they are evolutionarily conserved at the amino acid sequence level. A plot of the amino acid conservation of Pezizomycotina CPC1 sequences relative to N. crassa sequence is shown in Fig. 1K. A highly conserved region is present near the C terminus of CPC1 (residues 430 to 500), which corresponds to the α-helix of the bZIP DNA binding domain (Fig. 1K). Excluding this, there are few highly conserved stretches, but the N-terminal extensions are as well conserved as the mORF. We examined two conserved regions in the N-terminal extension (Fig. 1K, red dashes) more closely to determine if the conservation is at the amino acid or the nucleotide level and, if it is at the amino acid level, which reading frame showed the highest conservation (Fig. S3A and B). The first conserved region examined overlaps uORF2. The proportion of synonymous substitutions was much higher in the mORF frame than in uORF2 frame (Fig. S3A). For the 5 codons showing the highest amino acid conservation in the mORF frame (Fig. 1K, orange dashed bracket), the ratio of synonymous to nonsynonymous substitutions is particularly striking. The second conserved region examined comprises 7 codons starting 16 codons upstream of the mAUG. This region also shows a high proportion of synonymous substitutions in the mORF frame compared to the other frames (Fig. S3B), indicating that its conservation occurs because of selection in the mORF frame.
Regions upstream of mAUG show coding conservation in frame with the mORF. (A) Conservation in the N-terminal extension of N. crassa cpc-1. (i and ii) Nucleotide and codon alignments of the region corresponding to uORF2 of cpc-1 in N. crassa are shown in frame with the main ORF (frame 1 in Fig. 1B) (i) and in frame with uORF2 (frame 3 in Fig. 1B) (ii). (iii to v) A proline-rich region (residues 218 to 224, red dashed bracket in Fig. 1K) upstream of the cpc-1 mAUG shows high conservation in the mORF. Nucleotide and codon alignments of this region are shown for frame 1 (in frame with the mORF) (iii), frame 2 (iv), and frame 3 (v). In these alignments, consensus codons (and gaps) are highlighted in white, synonymous codons in green, nonsynonymous codons in brown, and stop codons in magenta. Species identifiers of the sequences used are indicated on the left. The numbers of synonymous and nonsynonymous substitutions are summed below each panel. The additional numbers in parentheses below panel i indicate the number of synonymous and nonsynonymous substitutions in frame with the mORF in the small patch of sequence marked by a dashed orange bracket above the aligned sequences. This patch represents the conserved peptide region indicated by the corresponding dashed orange bracket in Fig. 1K. The alignments were generated by a custom script that conceptually translated the main ORF frame into peptide sequence, then performed ClustalX alignment of peptide sequence, and then converted this alignment back to nucleotides. The most common codon in each column was then designated the consensus codon; synonymous and nonsynonymous substitutions were determined relative to it. (C) The region corresponding to the beginning of uORF3 of Coprinus cinerea cpc-1 is conserved at the amino acid level in frame with the mORF in Agaricomycetes homologs. Nucleotide and codon alignments of the region, including the beginning of uORF3 of C. cinerea cpc-1 (indicated by red dashed lines in Fig. S5F), are shown for frame 1 (in frame with the main ORF) (vi), frame 2 (in frame with uORF3) (vii), and frame 3 (viii). Species identifiers of the sequences used are indicated on the left. “Pinus_taeda_cont” indicates a sequence coming from an unknown Agaricomycetes species contaminating the Pinus taeda EST library. The numbers of synonymous and nonsynonymous substitutions are indicated on the right of each panel. The position of the AUG start codon of uORF3 is indicated by an inverted red triangle. Coloring scheme, alignment generation, and display are the same as in other panels. Download FIG S3, PDF file, 2.6 MB (2.7MB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The coding potential of the upstream extension was analyzed with MLOGD (35). MLOGD calculates coding potential by using the patterns of substitutions observed across a sequence alignment to compare a coding model with a noncoding model via a likelihood-ratio test. When applied in a 20-codon sliding window, MLOGD detected a positive coding signature within the CPC1 AUG-initiated ORF (as expected) and upstream throughout the extension as far 5′ as NCC6 (Fig. S4). The coding signature was weaker (but still positive) from NCC6 to around one-third of the way through uORF2. This may be a result of increased CPC1 frame synonymous site conservation in this region (Fig. S4), leading to fewer sequence variations for MLOGD to distinguish between the coding and noncoding models. The enhanced synonymous site conservation (Fig. S4) is indicative of overlapping functional elements putting extra constraints on sequence evolution in this region, likely including the initiation contexts of NCC6 to NCC8 and the overlapping uORF2. The ratio of nonsynonymous to synonymous substitutions, dN/dS, was calculated for the region between NCC8 and the CPC1 AUG using codonml (36) and found to be 0.348 + 0.033 and thus statistically significantly less than 1 (99% confidence interval, 0.26 to 0.43), indicating that the upstream extension is indeed subject to purifying selection at the amino acid level, consistent with its being a coding sequence. Since synonymous site conservation interferes with use of dS as a proxy for neutral evolution, we also calculated dN/dS for the region from 21 codons after NCC8 to the CPC1 AUG (Fig. S4), giving a dN/dS ratio of 0.285. For comparison, the dN/dS ratio for the region between the CPC1 AUG and CPC1 stop codon was found to be 0.144 + 0.012, indicating stronger purifying selection on average in the AUG-initiated CPC1 ORF than in the upstream extension. Taken together, these data indicate that the mRNA sequences specifying the N-terminal extension are under purifying selection in the mORF frame.
Coding potential and synonymous site conservation in cpc-1. At the top is a map of the N. crassa maximal CPC1-frame ORF region, showing the main CPC1 ORF; uNCC6, uNCC7, and uNCC8; and uORFs 1 and 2. Immediately below is shown the coding potential in the three reading frames as measured with MLOGD using an alignment of the maximal CPC1-frame ORF regions from 96 fungal species and a 20-codon sliding window. MLOGD uses a principle similar to the dN/dS statistic (ratio of nonsynonymous to synonymous substitutions) but also accounts for conservative amino acid substitutions being more probable than nonconservative substitutions. Positive scores indicate that the sequence is likely to be coding in the given reading frame and are observed throughout CPC1 and most of the upstream extension, at least from uNCC8. Positions where >50% of the alignment divergence is missing due to alignment gaps are omitted. Below this is shown alignment divergence (aln. divg, mean number of substitutions per nucleotide) for the sequences that contribute to the statistics at each position in the alignment (in any particular column, some sequences may be omitted from the statistical calculations due to alignment gaps). The bottom panels show the analysis of synonymous site conservation across the alignment. The brown line (“o/e”) indicates the ratio of the observed number of synonymous substitutions within a 5-codon window to the number expected under a null model of neutral evolution at synonymous sites, while the red line (“p-val”) depicts the corresponding statistical significance. Download FIG S4, PDF file, 0.1 MB (138.1KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We next investigated the architecture of cpc-1 homologs in fungi outside the Pezizomycotina. Examination of multiple sequences from other classes within the Ascomycota, including Saccharomycotina (including S. cerevisiae GCN4) and Taphrinomycotina, showed that these cpc-1 homologs lack the analogous N-terminal extensions of the main ORF. Thus, the conserved N-terminal extension in Ascomycota is confined to Pezizomycotina. Little comparative sequence information was available to examine other fungal phyla except for Basidiomycota. Within this phylum, analysis was complicated by the presence of multiple cpc-1 paralogs in some species. Typically, the 5′ leaders of cpc-1 homologs from Basidiomycota have 3 to 4 uAUGs. These can either initiate, or exist within, the reading frames of two or three uORFs (Fig. S5). uORF1 is 4 to 7 codons long, while one of the downstream uORFs, initiated by AUG in a good context, is much longer (uORFL). Crucially, uORFL sometimes overlaps the mORF (see Fig. S5B and C). Examination of 32 cpc-1 Basidiomycota mRNA sequences (3 from Ustilaginomycotina, 27 from Agaricomycetes, and 2 from Microbotryomycetes) with identifiable uORFs revealed that, in all cases, no stop codon in frame with the mORF is present between uORF1 and the mAUG (Fig. S5A and B). In fact, no stop codon in frame with the mORF is closer than 77 nucleotides upstream of uORF1. Unlike in Pezizomycotina, where three highly conserved NCCs were identified for most N-terminal extensions, no well-conserved NCCs were identified in Basidiomycota. However, in every Basidiomycota cpc-1 homolog, several NCCs in good initiation contexts and in the same frame with the mORF are present 5′ of the apparently inhibitory uORFL. In all cases, the first NCC is located 5′ of uORF1 such that potential translation initiation at the NCC would bypass the regulatory effects of the uORFs.
We searched the 27 uORF-containing cpc-1 homologs from Agaricomycetes for conserved features. In these, there is a single conserved NCC capable of initiating translation of an N-terminal extension and this NCC is present at least 31 nucleotides 5′ of the uORF1 AUG (Fig. S5B). Although the position of this NCC is well conserved, its identity is not. In most cases, it is AUU; in others, it is UUG, AUA, or CUG (Fig. S1B). The specific identities of these NCCs appear largely specific to phylogenetic branches.
Features of cpc-1 homologs from Basidiomycota. (A to C) Diagrams of the cpc-1 transcripts from different Basidiomycota branches. AUG codons (green bars), stop codons (red bars), mORFs (black bars), and uORFs (blue bars) are indicated. The numbers of sequences used for the comparisons are shown in parentheses. Features are drawn to scale. (A) Ustilago maydis (representing the three available Ustilaginomycotina sequences). Conserved NCCs in frame 1 upstream of uORF2 are indicated by magenta bars. (B) Coprinopsis cinerea (representing the 27 available Agaricomycetes sequences containing uORFs). The position of the single conserved NCC in frame 1 upstream of uORF3 is indicated by a magenta bar. (C) Leucosporidium scottii (representing the two available Microbotryomycetes sequences containing uORFs). The position of the single conserved NCC in frame 1 upstream of the uORF is indicated by a magenta bar. (D and E) Frequency WebLogo of the conservation of the initiation contexts, from −6 to +4, of all C. cinerea genes (D) and the single conserved NCC in frame 1 of Agaricomycetes upstream of the uORFs (E). (F) The amino acid sequence encoded by frame 1 of C. cinerea cpc-1 mRNA (the mORF), bounded by two in-frame stop codons. mAUG indicates the annotated initiation codon; the position of the conserved upstream NCC is also indicated. The approximate positions of uORF1, uORF2, and uORF3 are indicated by blue lines. The C-terminal bZIP domain is indicated in red font. The region analyzed in Fig. S3C is bracketed by a dashed red line. The conservation of each residue from the alignment of homologs from 27 species (those used to construct the tree in Fig. S1B) is shown below the amino acid sequence. Download FIG S5, PDF file, 0.3 MB (311KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The preferred initiation context in Agaricomycetes, as determined by analyses of Coprinopsis cinerea (Fig. S5D), is similar to the context in both Pezizomycotina and mammals. Based on this, the context of the single conserved NCC in the 5′ leaders of cpc-1 homologs in Agaricomycetes appears favorable if not optimal (Fig. S5E). The putative N-terminal extensions in Agaricomycetes are shorter than in Pezizomycotina—approximately 120 versus 180 amino acids, respectively. The amino acid conservation in Agaricomycetes is also concentrated in the C-terminal region of the mORF that contains the α-helix including the bZIP DNA binding domain (red letters in Fig. S5F). Patches of substantial conservation are observed within the 50 amino acids upstream of the mORF. The most highly conserved stretch in this region was subjected to a more careful examination (Fig. S3C, red dashed line). This sequence overlaps the last, and usually longest, uORF. This analysis indicates that conservation of amino acid sequence of the N-terminal extension in the mORF frame is more important than conservation in the uORF or the third reading frame (Fig. S3C), consistent with the findings in Pezizomycotina.
A peculiar mRNA architecture exists in cpc-1 homologs in Microbotryomycetes (Fig. S5C). Even though only two uORF-containing homologs of cpc-1 were obtained in this branch of Basidiomycota, both transcripts have the same unusual feature (Fig. S5C). Unlike Ustilaginomycotina (Fig. S5A) or Agaricomycetes (Fig. S5B), no evidence was detected for the existence of a short regulatory uORF1. The 5′ end of the homolog from Leucosporidium scottii is well supported by several expressed sequence tags (ESTs), and the upstream neighboring gene is in close proximity. Thus, instead of a uORF1, a long uORF initiated by AUG in a favorable initiation context is present, which overlaps the mORF. No in-frame stop codons are seen upstream of the mAUG. A single conserved NCC can be identified upstream of the uORF start codon so that ribosomes initiating from this NCC could synthesize an N-terminally extended CPC1 isoform and completely bypass any inhibitory effects of the uORF.
Experimental analyses of N. crassa cpc-1.
To investigate the effects of uORF1, uORF2, and upstream NCCs on the translation of N. crassa CPC1 in cell extracts, the 5′ leader of cpc-1, including the first two codons of the mORF, was fused in frame to firefly luciferase (cpc-1–luc, designated wild type [WT] [Fig. S6]). The functions of initiation codons identified by bioinformatics approaches were tested by mutational analyses of this construct. A UAA mutation (designated UAA) was introduced in frame with, and 12 nt upstream of, the mAUG to terminate translation and therefore truncate translation products that initiated from upstream NCCs. The start codon of uORF1 was mutated to AAA (ΔuORF1), that of uORF2 was mutated to ACA (ΔuORF2), and that of the mORF was mutated to CTC (ΔmAUG), to eliminate their initiation activity.
The sequences of cpc-1–luc fusion constructs used for in vitro experiments. Mutations and selected unique restriction sites are shown below the sequence. uORF1, uORF2, and the beginning of the main ORF (mORF) are shaded gray. NCCs are shaded magenta and are numbered 1 to 8. The position of the introduced TAA mutation that terminates the translation from NCCs is indicated by a red box. Luciferase was placed in frame with CPC1 at the XhoI site. Download FIG S6, PDF file, 0.2 MB (217.6KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The functions of uORF1 and uORF2 were examined by mutating their start codons separately or together. First, we examined these mutations in constructs containing the UAA mutation to look specifically at luciferase synthesis from the mAUG. Luciferase synthesis was measured by enzyme activity assay and by labeling with [35S]Met (Fig. 2A). Compared to a construct containing both uORFs, the ΔuORF1 mutation diminished translation of the mORF as indicated by a reduced level of luciferase activity (15%) and decreased production of [35S]Met-labeled polypeptides (compare constructs 1 and 2, Fig. 2A). The ΔuORF2 mutation increased translation from the mAUG approximately 2.9-fold (compare constructs 1 and 3, Fig. 2A). For the ΔuORF1 ΔuORF2 double mutant, the synthesis of luciferase increased (compare constructs 1 and 4, Fig. 2A), but this increase was less than for ΔuORF2 alone. These data suggest that reinitiation occurs after translation of uORF1, that translation of uORF2 is inhibitory, and that a fraction of ribosomes that translate uORF1 reinitiate at uORF2. In the absence of uORF1 and uORF2, synthesis of luciferase is lower than in the absence of uORF2 alone. This could be explained if the NCCs are used more efficiently in the absence of uORF1 (see below).
FIG 2 .
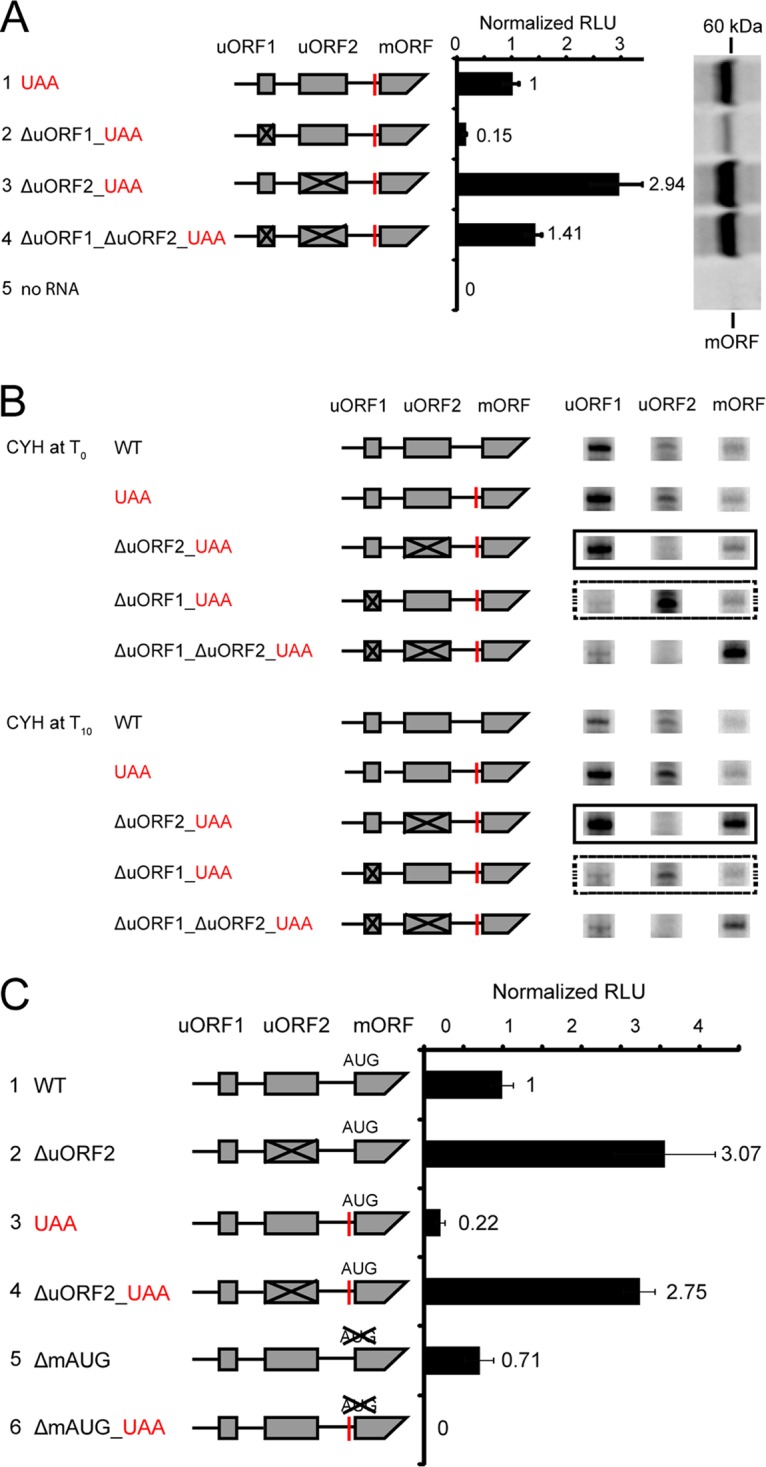
Contribution of cpc-1 uORF1 and uORF2 to the regulation of translation from the mAUG in N. crassa cell extracts. (A) Effects of eliminating cpc-1 uORF1 and uORF2 on translation from the mAUG. Constructs (numbered 1 to 4) contained the UAA stop codon (red bar) to eliminate translation from upstream in-frame NCCs and the indicated mutations to uORF start codons to eliminate initiation from them (uORF1 AUG to AAA and/or uORF2 AUG to ACA). Capped and polyadenylated mRNA (6 ng) was used to program N. crassa translation reaction mixtures (10 µl). LUC activity produced from mRNAs 2 to 4 obtained after 30 min of incubation at 26°C was calculated relative to the activity produced from mRNA 1. Mean values and standard deviations from three independent experiments, each performed in triplicate, are given as normalized relative light units (RLU). In addition, [35S]Met-labeled translation products from translation reactions programmed with mRNAs 1 to 4 or with no mRNA were analyzed on 12% NuPAGE gels, and a representative gel is shown. The position of radiolabeled LUC produced from the mAUG is indicated. (B) Toeprint analysis indicates reinitiation following translation of cpc-1 uORF1 but not uORF2. cpc-1–luc mRNA (60 ng) was used to program 20-µl N. crassa cell-free translation reaction mixtures. WT mRNA containing the wild-type cpc-1 5′ leader and the mRNAs used in panel A were analyzed in parallel along with controls. Reaction mixtures were incubated at 26°C min with cycloheximide (CYH) added either prior to incubation (T0) or after 10 min of incubation (T10) as indicated. Radiolabeled primer CPC101 was used to examine ribosomes at uORF1 and uORF2; primer ZW4 was used to examine ribosomes at the mORF. The original data from which the toeprint signals were excised are shown in Fig. S7. (C) Discriminating translation from N. crassa cpc-1 NCCs and mAUG in vitro. Capped and polyadenylated mRNA (6 ng) was used to program N. crassa translation reaction mixtures (10 µl) with the indicated constructs. Firefly luciferase activity from each mRNA obtained after 30 min of incubation at 26°C was calculated relative to synthesis from the WT construct. Mean values and standard deviations from three independent experiments, each performed in triplicate, are plotted.
In earlier studies on S. cerevisiae GCN4, we used toeprint analyses to demonstrate reinitiation following uORF1 but not uORF4 translation in S. cerevisiae extracts (12). We adapted a similar approach to examine cpc-1 uORF1 and uORF2 in N. crassa extracts. Adding cycloheximide (CYH) to reaction mixtures at time zero (T0) allows toeprint mapping of initiation codons where 80S ribosomes first initiate translation following initial scanning. Adding CYH at 10 min of incubation of translation reaction mixtures (T10) allows toeprint mapping of initiation sites in the steady state, for example, at additional sites where ribosomes have reinitiated. At T0 and T10, ribosomes are seen at the uORF1 AUG start codon; mutation to AAA eliminated this signal (Fig. 2B). This is expected since the uORF1 AUG is in an optimal initiation context. At T0, a reduced toeprint signal is seen at the uORF2 AUG relative to the signal at the uORF1 AUG. When the uORF1 AUG is mutated, the uORF2 AUG signal increased substantially; mutation of the uORF2 AUG to ACA eliminated this signal. These data indicate that most ribosomes initiate at uORF1 but, when it is absent, they scan to uORF2. At T0, a relatively low signal was observed at the mORF AUG except when uORF1 and uORF2 AUGs were mutated, as expected from scanning. When CYH was added at T10, the most dramatic change in signal was an increase at the mAUG in the ΔuORF2 construct. This increase of the mAUG was not seen in the ΔuORF1 construct or the ΔuORF1 ΔuORF2 construct. These data are consistent with ribosomes reinitiating at the mAUG following uORF1 translation in vitro. They suggest that uORF1 and uORF2 of N. crassa cpc-1 function similarly to uORF1 and uORF4, respectively, of S. cerevisiae GCN4.
We next compared luciferase activities obtained from constructs with and without the introduced in-frame UAA stop codon to examine translation from NCCs upstream of the mAUG (Fig. 2C). The production of luciferase decreased when the UAA was present, indicating that polypeptides with luciferase activity were produced using NCCs upstream of the mORF (compare constructs 1 and 3, constructs 2 and 4, and constructs 3 and 6 in Fig. 2C). The UAA mutation decreased luciferase synthesis in the presence or absence of uORF2 (compare constructs 1 and 2 and constructs 3 and 4 [Fig. 2C]). Elimination of uORF2 resulted in overall increased luciferase synthesis as expected from its proposed inhibitory role for initiation at mAUG. As expected, NCCs and the mAUG have separate roles in initiation; combining ΔmAUG and UAA mutations yielded no detectable luciferase (Fig. 2C).
Interestingly, ΔmAUG showed a relatively small decrease in luciferase activity compared to WT (ΔmAUG, 71% of WT; compare constructs 1 and 5, Fig. 2C). This observation is consistent with the differences between the WT and the UAA constructs (UAA, 22% of WT; compare constructs 1 and 3, Fig. 2C). These data indicate that upstream NCCs are used to initiate translation efficiently in vitro. While this is the case, we did not identify NCCs by toeprint analyses (Fig. S7). Possibly, translation initiation is distributed among multiple NCCs, reducing signals at individual NCCs.
Toeprint analysis indicates reinitiation following translation of cpc-1 uORF1 but not uORF2 (primary data for Fig. 2B). The description of the experiment is given in the text. For markers, either primer CPC101 (upper panel) or ZW4 (lower panel) was used to sequence the WT template (the four leftmost lanes). The nucleotide complementary to the dideoxynucleotide added to each sequencing reaction for the wild-type cpc-1–luc template is indicated above the corresponding lane so that the sequence of the template can be directly deduced; the 5′-to-3′ sequence reads from top to bottom. Asterisks (top to bottom), toeprint products corresponding to ribosomes bound at uAUG1, uAUG2, and mAUG on the cpc-1–luc RNA. Boxes (top to bottom), uAUG1, uAUG2, and mAUG in the sequence markers. The ribosome protects approximately 16 nt 3′ of a start codon in the ribosome P-site. Download FIG S7, PDF file, 0.3 MB (287.4KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The eight NCCs identified bioinformatically (Fig. S6) were individually eliminated, and the consequences were examined by [35S]Met labeling in N. crassa and wheat germ extracts (Fig. 3 and S7). These mutations were also combined with the UAA mutation so that the resulting polypeptides produced from upstream initiation could be better resolved by SDS-PAGE. Elimination of NCC1, NCC2, NCC3, or NCC4 did not yield any detectable differences compared to UAA (Fig. S8, lanes 3 to 7). In contrast, elimination of NCC5, -6, -7, or -8 resulted in disappearance of specific truncated polypeptides (Fig. S8, lanes 8 to 11 and 3, and Fig. 3, lanes 8 to 11 and 3), indicating that NCC5 to NCC8 initiated translation in N. crassa and wheat germ systems. When NCC8 (ACG) was changed to AUG, the signal in the corresponding band increased as expected (lanes 12 and 3, Fig. S8, and lanes 8 and 3, Fig. 3).
FIG 3 .

Evidence that N. crassa NCC5 to NCC8 initiate translation in vitro. Synthetic RNAs (60 ng) for the indicated constructs were used to program 10 µl of cell-free translation systems from N. crassa (left) or wheat germ (right). Reaction mixtures were incubated for 30 min at 26°C. Radiolabeled products were analyzed on 12% NuPAGE gels. Open circles, translation products eliminated upon mutation of NCC5 to NCC8; the product predicted to be initiated from NCC8 also increased when NCC8 was changed to AUG (lane 8). Arrowhead, position of mAUG-initiated translation product (mORF). Brackets, translation products larger than the mORF produced in the absence of an in-frame UAA stop codon.
Evidence from [35S]Met labeling showing that NCC5 to -8, but not NCC1 to -4, initiate translation in an N. crassa cell-free system. Synthetic RNAs (60 ng) for the indicated constructs were used to program 10 µl of cell-free translation reaction mixtures from N. crassa. Reaction mixtures were incubated for 30 min at 26°C. Radiolabeled products were analyzed on 12% NuPAGE gels. Open circles, translation products eliminated upon mutation of NCC5 to -8; the product predicted to be initiated from NCC8 also increased when NCC8 was changed to AUG (lane 12). Arrowhead, position of mAUG-initiated translation product (mORF). Brackets, translation products larger than the mORF produced in the absence of an in-frame UAA stop codon. Download FIG S8, PDF file, 0.1 MB (134KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cell translation extracts programmed with CPC1-LUC (WT) produce polypeptides migrating more slowly than luciferase synthesized from an mRNA specifying LUC alone or a CPC1-LUC mRNA with the UAA mutation (Fig. 1A, 3, and S8). When NCC5, NCC6, NCC7, and NCC8 were eliminated together in the absence of the UAA mutation, polypeptides larger than luciferase were still observed, although the amount was reduced compared to WT (Fig. 3, compare lanes 10, 11, and 3). This suggests that other upstream codons in the cpc-1 upstream region can be used to initiate polypeptide synthesis. This was observed in the presence or absence of uORF1 (lanes 10 and 11, Fig. 3).
To examine the roles of upstream NCCs in translation in vivo in N. crassa, strains containing N. crassa codon-optimized luciferase fused in frame with wild-type or mutated cpc-1 5′ sequences were constructed. Three independent transformants containing each construct were used to measure LUC activity and LUC mRNA levels. We examined WT, UAA, and ΔmAUG strains and the ΔmAUG UAA double mutant. Luciferase activity was measured and normalized to reporter mRNA levels to account for the small differences in luciferase mRNA levels observed. Expression levels of WT and UAA reporters were similar (in Fig. 4). Luciferase activity from the ΔmAUG construct was much lower, but this activity was higher than that for the ΔmAUG UAA construct. For the ΔmAUG construct, higher luciferase activity was observed than for the ΔmAUG UAA double mutant. Thus, although the amount of luciferase activity derived from upstream NCCs was less than 1% of activity from the mAUG in vivo, detectable luciferase was nevertheless observed (compare constructs 3, 1, and 2 in Fig. 4 and compare constructs 5, 1, and 3 in Fig. 2C). Possibly, NCCs were not used as efficiently in vivo as in vitro. Alternatively, N-terminally extended luciferases are less stable or less active in vivo, but we have not investigated this further.
FIG 4 .

Discriminating translation from N. crassa cpc-1 NCCs and mAUG in vivo. Constructs 1 to 4 were placed at the N. crassa his-3 locus (three independent transformants of each). LUC activities were measured, and values were plotted relative to WT. Black bars, LUC activities normalized to total extracted protein; gray bars, LUC activities normalized first to total protein and then to luc mRNA/cox-5 mRNA levels. Mean values and standard deviations for all measurements are derived from three independent experiments, each using all independent transformants.
For further investigation of translational activity in the region of cpc-1 mRNA upstream of mAUG, data from ribosome profiling experiments in N. crassa were examined. Ribosome profiling provides snapshots of genome-wide in vivo translation by deep sequencing which is amenable to quantification. Cells were grown for 24 h in the dark, and ribosome profiling data were collected and analyzed as described in Materials and Methods. As shown previously, ribosome footprint data can be used to determine the frame in which a particular region of mRNA is being translated (29, 37, 38). The ribosome footprints for the cpc-1 transcript (Fig. 5) show that uORF1 and uORF2 are heavily translated under these conditions. The frame information obtained with protected fragments of at least 28 nt using a 15-nt offset to the ribosome A site agrees with the predictions that, relative to the CPC1 frame, uORF1 is in frame 2 and uORF2 is in frame 3. The main CPC1 coding region contains ribosomes in the predicted reading frame (frame 1) as expected. Importantly, ribosome footprints in the 5′ region of the transcript outside uORF1, uORF2, and CPC1, especially between uORF2 and CPC1, were all in the CPC1 frame. This is consistent with the in vitro data showing in-frame translation upstream of the main CPC1 coding region. Furthermore, ribosome footprint data, with certain preparation protocols, show accumulation of footprints at AUG and non-AUG start codons (20, 29). This is also the case in the data set that was used for the present analysis of N. crassa. Accumulated footprints were observed at uAUG1 and uAUG2 and at four of the eight predicted NCCs (Fig. 5).
FIG 5 .

Ribosome profiling evidence for translation of the cpc-1 N-terminal extension. The ribosome-protected fragment count (cutoff at 50) for each position 5′ to 3′ along the cpc-1 mRNA is shown on top. Peaks that can be assigned to specific initiation codons, as labeled in Fig. 1B and K, are indicated in red. A schematic map of the ORF organization of cpc-1, drawn to scale, is shown below the ribosome-protected fragment count. The uORFs are represented by blue rectangles. The eight in-frame NCCs upstream of uORF2 are represented by magenta bars. The mORF is represented by a black rectangle. The reading frame of each feature relative to the mORF is indicated. The reading frame information derived from the ribosome-protected fragments for 5 specific regions along the cpc-1 mRNA, as well as the tabulated data for all N. crassa coding sequences (CDSs), is shown at the bottom. The tabulation is done by scoring the first nucleotide of each ribosome-protected fragment relative to the reading frame of the annotated coding sequence (first to last ribosome A-site codons), using a 15-nucleotide 5′ offset—the proportions of total fragments mapped to frame 1 are shown by green bars, those mapped to frame 2 are shown by red bars, and those mapped to frame 3 are shown by yellow bars. The number of reads used to tabulate the data in each histogram is indicated as “N.”
As controls, we examined ribosomes in the 5′ leaders of arg-2 (NCU07732) and eif5 (NCU00366), which are two other N. crassa mRNA transcripts that contain uORFs (31, 39). In addition to the ribosome footprints in the main ORFs that preferentially corresponded to the predicted reading frame, ribosomes are observed in the uORFs (Fig. S9). The frame information of the footprints in the main ORFs of the uORFs matches the predictions based on the gene model of the corresponding mRNAs. The footprints in the 5′ leaders outside the uORFs appear not to be spurious. Positions represented by more than 2 footprints correspond to near-cognate start codons. For example, each of the six larger peaks 5′ of the first uORF of eIF5 precisely (i.e., with 1-nt resolution) matches six NCCs—AUC, UUG, CUG, GUG, UUG, and UUG, respectively.
Ribosome profiling data, including framing information, for arg-2 and eif5. The ribosome-protected fragment count for each position 5′ to 3′ along the mRNA is shown on top for arg-2 (NCU07732) (A) and eif5 (NCU00366) (B). Peaks corresponding to specific NCCs in the 5′ leader of eif5 are shown by red arrows. A schematic map of the ORF organization, drawn to scale, is shown below the ribosome-protected fragment count graph. The reading frame information derived from the ribosome-protected fragments for specific regions along the mRNAs is shown below the schematic maps. AAP, uORF-encoded arginine attenuator peptide; all CDSs, all predicted N. crassa genic ORFs. The color coding for different components of the figure is the same as in Fig. 5. Download FIG S9, PDF file, 0.2 MB (252.8KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Although the ribosome profiling experiment described above provided strong evidence for in vivo translation of the region upstream of the mAUG in the CPC1 frame, it did not and could not address the question of whether translation of this region increases or decreases under stress conditions. The answer is important to address the question of whether the N-terminal extension is part of the translational regulation of cpc-1 or whether it merely provides a constitutive alternative isoform of cpc-1. To address this question, a second ribosome profiling experiment was performed. In it, N. crassa cells were grown in the presence or absence of 3-amino-1,2,4-triazole (3AT), which induces histidine starvation. Compared to untreated cells, as expected, 3AT cells showed elevated density in the mORF compared to uORF2, with the ratio of the ribosome footprint counts in the two regions changing from 0.57 in untreated cells to 1.66 in 3AT-treated cells. However, the ratio of the ribosome footprint count in the region between uORF2 and the mORF relative to the footprint count in the mORF remains nearly unchanged—0.19 in untreated cells to 0.21 in 3AT-treated cells. This result is consistent with the idea that amino acid starvation induces translation at the non-AUG codons of cpc-1 responsible for initiation of the N-terminal extension. Consistent with this notion, the ratio of ribosome footprint count in the region between uORF1 and uORF2, where non-AUG initiation of the N-terminal extension must occur, to the footprint count in uORF2 increases in 3AT-treated cells compared to control cells—from 0.11 to 0.24.
DISCUSSION
We examined the structures of N. crassa cpc-1 homologs in fungi for which sequence was available. In Pezizomycotina, all cpc-1 genes specify two uORFs, uORF1 and uORF2, within an extended mRNA 5′ leader. The data obtained here with N. crassa are consistent with uORF1 and uORF2 functioning analogously to S. cerevisiae GCN4 uORF1 and uORF4, respectively, to control initiation at the predicted mAUG start codon. Surprisingly, a long, conserved coding region upstream of this AUG start codon that was in frame with CPC1 was present in all homologs from Pezizomycotina, and in some cases, this open reading frame extended to the predicted mRNA 5′ ends. While no AUG codons were observed that could produce N-terminally extended isoforms of CPC1 (excepting the possibility of ribosomal frameshifting from a uORF AUG), near-cognate start codons (NCCs), some well conserved, were present in the CPC1 reading frame that potentially could initiate translation of such isoforms. Translation initiating from four conserved NCCs in the N. crassa cpc-1 5′ leader was observed in vitro in N. crassa and wheat germ translation extracts. Utilization of NCCs in vivo would result in synthesis of alternative isoforms of CPC1; these isoforms may have similar or different functions than CPC1 produced from the main AUG. N-terminal extensions could also influence protein stability. Only future experiments can distinguish between these possibilities. The synthesis of these alternative isoforms from NCCs upstream of uORF2 would also bypass the inhibitory effect of uORF2, which reduces synthesis of CPC1 from the downstream main AUG. These findings suggest a model for additional translational regulation of Pezizomycotina cpc-1 through the use of NCCs, which could be independent of the uORF control model elucidated for S. cerevisiae GCN4. Another potential mechanism that could contribute to translation in the CPC1 reading frame upstream of the main AUG that is also consistent with these data is +1 (or −2) translational frameshifting occurring within uORF2, since all uORF2s in Pezizomycotina analyzed thus far are in the −1 frame relative to the mORF.
We found no fungal homologs of cpc-1/GCN4 outside Pezizomycotina and Basidiomycota that have NCC-initiated N-terminal extensions with the potential to preempt the effect of translating a long and inhibitory uORF. Since the other two subphyla of Ascomycota, Saccharomycotina and Taphrinomycotina, do not have potential for NCC-initiated extensions, it not entirely clear if the conserved extensions present in Pezizomycotina and in Basidiomycota (a sister phylum of Ascomycota in the subkingdom Dikarya) are homologous and were present in the last common ancestor of Dikarya, which lived around 500 million years ago (33), or whether they are examples of convergent evolution.
In the studies reported here, there is a discrepancy between luciferase activities produced in vitro and those produced in vivo from the N. crassa cpc-1 NCCs. At face value, this would mean that in vitro there is more initiation from NCCs than from the mAUG, while in vivo the situation is reversed. For the in vitro experiments, we used an intermediate [Mg2+], which favors AUG over NCC initiation, but the in vitro conditions used here are not expected to be as stringent as in vivo (17). Thus, we expect that relatively more NCC-initiated products would be produced in vitro than in vivo, but the discrepancy in levels of CPC1-LUC activity observed still seems too large to be simply accounted for by this consideration, given that the ribosome profiling data support translation from NCCs in vivo. It is possible that the N-terminally extended forms of the luciferase reporter are unstable in vivo and that luciferase reporter data might thus provide accurate information on the relative level of N-terminally extended CPC1 isoforms in vivo. This level, while low, could nevertheless be physiologically significant. This conclusion is further strengthened by the ribosome profiling data. It too suggests that under normal conditions translation of the mORF, though low, is primarily initiated upstream of the stop codon of uORF2 (e.g., at NCCs). Taken together, these data raise important new questions regarding the functions of the isoforms of CPC1 and their regulation.
The results from ribosome profiling following 3AT treatment raise several intriguing questions regarding the likelihood that the NCCs in cpc-1 are used for regulation and also about the nature of this regulation. The standard model of cpc-1 regulation under amino acid limitation posits that eIF2 phosphorylation reduces translation of the inhibitory uORF2 by lengthening the time of reinitiation. Yet, total translation of the region between uORF1 and uORF2 appears to increase following 3AT treatment. Either NCC initiation becomes very efficient under amino acid limitation in general, overcoming the inhibitory effect of reduced reinitiation, or translation of uORF1 specifically primes retained ribosomes for initiation at the NCC in response to amino acid limitation.
CPC1 is a bZIP transcription factor, and the mammalian bZIP transcription factor family of CCAAT/enhancer binding proteins (C/EBPs) provides potential context for how bZIP isoforms are produced by alternative initiation to have different functions. C/EBPα initiates at an in-frame AUG in a poor initiation context; C/EBPβ initiates from an NCC in a good context (40). Leaky scanning past the latter initiation codon leads to initiation at an out-of-frame AUG codon in good context, producing a short ORF. Two additional C/EBP isoforms (LAP and LIP) are generated by initiation from in-frame AUG codons downstream of this short ORF by reinitiation following translation of the short ORF, and the relative levels of LAP and LIP can be altered by changes in eIF2α phosphorylation. LAP functions as a transcriptional activator and LIP functions as a transcriptional repressor, modulating different transcriptional outcomes under “normal” and stress conditions.
It is worth considering that the translation of another fungal bZIP transcription factor, Podospora anserina IDI-4, is proposed to initiate from a CUG and not an AUG codon, and this CUG is conserved in the N. crassa homolog (41). Thus, possibly, fungal bZIP transcriptional factors may more generally use NCCs to initiate their translation.
The physiological conditions that govern initiation at NCCs are an emerging area of investigation, and the evolutionarily conserved features in the 5′ UTRs of filamentous fungal CPC1 homologs provide an additional new architecture to confer 5′ UTR translation regulation (42). In S. cerevisiae, amino acid limitation increases initiation at NCCs (29), as does the shift to the meiotic developmental program (43), at least for genes other than GCN4. A chemical screen identified several compounds that increase the efficiency of initiation at NCCs (44). The concentration of free polyamines affects initiation from a conserved AUU start codon of a uORF within the mRNA encoding AZIN1 in mammalian cells (45). A number of cellular factors are known to be involved in discrimination between favorable and unfavorable initiation codons and contexts (46–49). Changes in the activity or cellular levels of eIF1 or eIF5 can have profound effects on translation initiation at NCCs or AUG codons in a poor context (9, 30, 31, 50–52). Understanding the physiological conditions that control initiation at NCCs has broad implication for gene regulation and protein synthesis as well as for specific understanding of these aspects of CPC1.
MATERIALS AND METHODS
Sequence assembly and analysis.
All cpc-1 sequences were obtained from GenBank by BLAST with the N. crassa cpc-1 sequence as the starting point. In most cases, the sequences were derived from the whole-genome shotgun contigs (WGS) database. WGS sequences were processed manually to predict intron/exon junctions for the mRNA sequence. In a minority of cases, sequences were available from expressed sequence tags (ESTs). EST data were manually assembled into contigs. Additional sequences were obtained from the transcriptome shotgun assembly (TSA) database. All alignments in this study were performed with the ClustalX2 and ClustalW algorithms. Sequences used in this study are available upon request.
Maximal (stop-codon-to-stop-codon) cpc-1 ORFs from 96 fungal species were translated and aligned as amino acids with MUSCLE (53), and the amino acid alignment was used to guide a codon-based nucleotide alignment (EMBOSS tranalign [54]). The alignment was mapped to N. crassa coordinates by removing all alignment columns that contained a gap character in the N. crassa sequence and analyzed with the codonml program in the PAML package (36), synplot2 (55) using a 5-codon sliding window, and MLOGD (35) using a 20-codon sliding window and 1-codon step size. For MLOGD, the null model in each window is that the sequence is noncoding while the alternative model is that the sequence is coding in the given reading frame. Standard deviations for the codonml dN/dS values were estimated via a bootstrapping procedure, in which codon columns of the alignment were randomly resampled (with replacement); 100 randomized alignments were generated for each region, and their dN/dS values were calculated with codonml.
Plasmids.
The starting point for all constructs was plasmid pPC01 (Z. Wang and M. Sachs, unpublished data), which has the 5′ leader of N. crassa cpc-1 cloned between BamHI and XhoI sites (the latter located at the 5′ end of the firefly luciferase cassette). First, the sequence GTCTTC, just upstream of the NCC8 ACG codon in the 5′ leader, was changed by two-step PCR to a SacI GAGCTC sequence to facilitate making subsequent mutations. This derivative is named pPC100 and is referred to as wild type (WT).
Specifics about plasmids are provided in Table S1A and B in the supplemental material. For in vitro experiments, pPC-series plasmids with the luciferase gene (not codon optimized) were used. When two PCR primers are shown in a cell in Table S1A, one-step PCR was used to generate inserted regions from corresponding PCR templates. When four PCR primers are shown, two-step PCR was used to generate inserted regions. PCR products and vectors were digested by restriction enzymes, gel purified, and ligated. For pPC176, synthetic complementary oligonucleotides were annealed and ligated to gel-purified vector pPC100 that had been digested with AgeI and XhoI. For in vivo assay mixtures that contained codon-optimized luciferase, plasmids pJI500, pJI502, pJI501, and pJI576 were made by replacing the small BamHI-NsiI fragment of pJI401 with the small BamHI-NsiI fragments of pPC100, pPC102, pPC101, and pPC176, respectively.
Plasmids (A) and primers (B) used in the study. Download TABLE S1, PDF file, 0.1 MB (101.9KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RNA synthesis and cell-free translation.
Capped and polyadenylated RNAs were transcribed in vitro by T7 RNA polymerase from plasmid DNA templates that were linearized with EcoRI, and the relative amounts of RNA were determined as described previously (17). In vitro translation and gel analysis for visualizing 35Met-labeled proteins using N. crassa extracts and wheat germ extract were accomplished as described previously (17), except that 10 µl of translation reaction mixtures was incubated for 30 min at 25°C and samples were mixed with 10 μl 2× NuPAGE LDS sample buffer (Invitrogen) and put on ice to stop reactions. In vitro translation for luciferase activity assays using N. crassa extracts was accomplished as described previously (17) using 6 ng of each mRNA to program extracts. Primer extension inhibition (toeprint) assays were accomplished using 32P-labeled primers CPC101 and ZW4 as described previously (17), except that 0.5 mg/ml cycloheximide was added to the reaction mixtures as indicated in Results.
Strains, culture conditions, and in vivo measurements.
Strain FGSC 6103 [his-3 (Y234M723) mat A] and the wild-type (WT) reference strain FGSC 2489 (74-OR23-1V mat A) were obtained from the Fungal Genetics Stock Center (FGSC) (56). Targeting of firefly luciferase reporters to the N. crassa his-3 locus by transformation of FGSC 6103 with PciI-linearized plasmid DNA (pJI500, pJI501, pJI502, or pJI576), culture conditions, and conditions for luciferase assays were as described previously (17). Total RNA was prepared from cells, cDNA was prepared, and reverse transcription-quantitative PCR (RT-qPCR) was performed as described previously (17).
Ribosome profiling.
N. crassa cultures were grown for 24 h in the dark, and following the breakage procedure used for the preparation of N. crassa cell translation extracts (57), but using the buffers described previously (58). Ribosome-protected mRNA fragments were prepared for sequencing essentially as described previously (58), except that 50 A260 units of lysate was used, nucleic acid pellets recovered from each step were washed with 80% ethanol, and the rRNA depletion step was omitted. Libraries were sequenced on an Illumina HiSeq 2000 sequencer, generating 54,446,346 51-mer reads after removal of multiplexing adapter sequences. Reads were further trimmed to remove the CTGTAGGCACCATCAAT adapter sequence with cutadapt-1.2.1 (options -n 1 -m 28) (59). Trimmed reads of 28 to 31 nt in length were mapped to Neurospora transcripts with Bowtie version 0.12.9 (options -n 0 -l 25 -a –norc) (60). Counts of reads at each framing position were generated with Python scripts as described previously (38).
Accession number(s).
Sequences of ribosome profiling (Ribo-Seq) libraries have been deposited in the NCBI Genome Expression Omnibus (GEO) under accession number GSE97717.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants (GM068087 to J.C.D., D.B.-P., M.F., and M.S.S. and GM47498 to M.S.S.), the Science Foundation Ireland (grant 08/IN.1/B1889 to J.F.A.), the Wellcome Trust (grant 106207) to A.E.F., and the Texas A&M Institute for Advanced Study (to J.C.D., D.B.-P., and M.S.S.). Funding for the open access charge was from the National Institutes of Health.
Footnotes
Citation Ivanov IP, Wei J, Caster SZ, Smith KM, Michel AM, Zhang Y, Firth AE, Freitag M, Dunlap JC, Bell-Pedersen D, Atkins JF, Sachs MS. 2017. Translation initiation from conserved non-AUG codons provides additional layers of regulation and coding capacity. mBio 8:e00844-17. https://doi.org/10.1128/mBio.00844-17.
REFERENCES
- 1.Hinnebusch AG. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 2.Hinnebusch AG, Natarajan K. 2002. Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell 1:22–32. doi: 10.1128/EC.01.1.22-32.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sachs MS. 1996. General and cross-pathway controls of amino acid biosynthesis, p 315–345. In Brambl R, Marzluf GA (ed), The Mycota: biochemistry and molecular biology, vol III Springer-Verlag, Heidelberg, Germany. [Google Scholar]
- 4.Kuo MH, vom Baur E, Struhl K, Allis CD. 2000. Gcn4 activator targets Gcn5 histone acetyltransferase to specific promoters independently of transcription. Mol Cell 6:1309–1320. doi: 10.1016/S1097-2765(00)00129-5. [DOI] [PubMed] [Google Scholar]
- 5.Tian C, Kasuga T, Sachs MS, Glass NL. 2007. Transcriptional profiling of cross pathway control in Neurospora crassa and comparative analysis of the Gcn4 and CPC1 regulons. Eukaryot Cell 6:1018–1029. doi: 10.1128/EC.00078-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ebbole DJ, Paluh JL, Plamann M, Sachs MS, Yanofsky C. 1991. cpc-1, the general regulatory gene for genes of amino acid biosynthesis in Neurospora crassa, is differentially expressed during the asexual life cycle. Mol Cell Biol 11:928–934. doi: 10.1128/MCB.11.2.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann B, Valerius O, Andermann M, Braus GH. 2001. Transcriptional autoregulation and inhibition of mRNA translation of amino acid regulator gene CPCA of filamentous fungus Aspergillus nidulans. Mol Biol Cell 12:2846–2857. doi: 10.1091/mbc.12.9.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tournu H, Tripathi G, Bertram G, Macaskill S, Mavor A, Walker L, Odds FC, Gow NA, Brown AJ. 2005. Global role of the protein kinase Gcn2 in the human pathogen Candida albicans. Eukaryot Cell 4:1687–1696. doi: 10.1128/EC.4.10.1687-1696.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinnebusch AG. 2011. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 75:434-467. doi: 10.1128/MMBR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinnebusch AG, Dever TE, Asano K. 2007. Mechanism of translation initiation in the yeast Saccharomyces cerevisiae, p 225–268. In Mathews MB, Sonenberg N, Hershey JWB (ed), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 11.Hinnebusch AG. 1988. Novel mechanisms of translational control in Saccharomyces cerevisiae. Trends Genet 4:169–174. doi: 10.1016/0168-9525(88)90023-6. [DOI] [PubMed] [Google Scholar]
- 12.Gaba A, Wang Z, Krishnamoorthy T, Hinnebusch AG, Sachs MS. 2001. Physical evidence for distinct mechanisms of translational control by upstream open reading frames. EMBO J 20:6453–6463. doi: 10.1093/emboj/20.22.6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu PD, Harding HP, Ron D. 2004. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vattem KM, Wek RC. 2004. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Z, Freitag M, Sachs MS. 1995. Translational regulation in response to changes in amino acid availability in Neurospora crassa. Mol Cell Biol 15:5235–5245. doi: 10.1128/MCB.15.10.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sattlegger E, Hinnebusch AG, Barthelmess IB. 1998. cpc-3, the Neurospora crassa homologue of yeast GCN2, encodes a polypeptide with juxtaposed eIF2α kinase and histidyl-tRNA synthetase-related domains required for general amino acid control. J Biol Chem 273:20404–20416. doi: 10.1074/jbc.273.32.20404. [DOI] [PubMed] [Google Scholar]
- 17.Wei J, Zhang Y, Ivanov IP, Sachs MS. 2013. The stringency of start codon selection in the filamentous fungus Neurospora crassa. J Biol Chem 288:9549–9562. doi: 10.1074/jbc.M112.447177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozak M. 1989. Context effects and inefficient initiation at non-AUG codons in eukaryotic cell-free translation systems. Mol Cell Biol 9:5073–5080. doi: 10.1128/MCB.9.11.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peabody DS. 1989. Translation initiation at non-AUG triplets in mammalian cells. J Biol Chem 264:5031–5035. [PubMed] [Google Scholar]
- 20.Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S, Liu B, Lee S, Huang SX, Shen B, Qian SB. 2012. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J, Brosch M. 2012. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res 22:2208–2218. doi: 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalstrup T, Blunck R. 2015. Reinitiation at non-canonical start codons leads to leak expression when incorporating unnatural amino acids. Sci Rep 5:11866. doi: 10.1038/srep11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanov IP, Firth AE, Michel AM, Atkins JF, Baranov PV. 2011. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res 39:4220–4234. doi: 10.1093/nar/gkr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, Vagner S. 2003. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol Cell 95:169–178. doi: 10.1016/S0248-4900(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 26.Firth AE, Brierley I. 2012. Non-canonical translation in RNA viruses. J Gen Virol 93:1385–1409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Damme P, Gawron D, Van Criekinge W, Menschaert G. 2014. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol Cell Proteomics 13:1245–1261. doi: 10.1074/mcp.M113.036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tzani I, Ivanov IP, Andreev DE, Dmitriev RI, Dean KA, Baranov PV, Atkins JF, Loughran G. 2016. Systematic analysis of the PTEN 5′ leader identifies a major AUU initiated proteoform. Open Biol 6:150203. doi: 10.1098/rsob.150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivanov IP, Loughran G, Sachs MS, Atkins JF. 2010. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc Natl Acad Sci U S A 107:18056–18060. doi: 10.1073/pnas.1009269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loughran G, Sachs MS, Atkins JF, Ivanov IP. 2012. Stringency of start codon selection modulates autoregulation of translation initiation factor eIF5. Nucleic Acids Res 40:2898–2906. doi: 10.1093/nar/gkr1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andreev DE, O’Connor PB, Zhdanov AV, Dmitriev RI, Shatsky IN, Papkovsky DB, Baranov PV. 2015. Oxygen and glucose deprivation induces widespread alterations in mRNA translation within 20 minutes. Genome Biol 16:90. doi: 10.1186/s13059-015-0651-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lücking R, Huhndorf S, Pfister DH, Plata ER, Lumbsch HT. 2009. Fungi evolved right on track. Mycologia 101:810–822. doi: 10.3852/09-016. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa S, Niimura Y, Gojobori T, Tanaka H, Miura K. 2008. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res 36:861–871. doi: 10.1093/nar/gkm1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Firth AE, Brown CM. 2006. Detecting overlapping coding sequences in virus genomes. BMC Bioinformatics 7:75. doi: 10.1186/1471-2105-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 37.Michel AM, Choudhury KR, Firth AE, Ingolia NT, Atkins JF, Baranov PV. 2012. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res 22:2219–2229. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei J, Wu C, Sachs MS. 2012. The arginine attenuator peptide interferes with the ribosome peptidyl transferase center. Mol Cell Biol 32:2396–2406. doi: 10.1128/MCB.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calkhoven CF, Müller C, Leutz A. 2000. Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev 14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- 41.Dementhon K, Saupe SJ, Clavé C. 2004. Characterization of IDI-4, a bZIP transcription factor inducing autophagy and cell death in the fungus Podospora anserina. Mol Microbiol 53:1625–1640. doi: 10.1111/j.1365-2958.2004.04235.x. [DOI] [PubMed] [Google Scholar]
- 42.Hinnebusch AG, Ivanov IP, Sonenberg N. 2016. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352:1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335:552–557. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takacs JE, Neary TB, Ingolia NT, Saini AK, Martin-Marcos P, Pelletier J, Hinnebusch AG, Lorsch JR. 2011. Identification of compounds that decrease the fidelity of start codon recognition by the eukaryotic translational machinery. RNA 17:439–452. doi: 10.1261/rna.2475211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivanov IP, Loughran G, Atkins JF. 2008. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc Natl Acad Sci U S A 105:10079–10084. doi: 10.1073/pnas.0801590105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pestova TV, Kolupaeva VG. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16:2906–2922. doi: 10.1101/gad.1020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donahue TF. 2000. Genetic approaches to translation initiation in Saccharomyces cerevisiae, p 595–614. In Sonenberg N, Hershey JWB, Mathews MB (ed), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 48.Nanda JS, Cheung YN, Takacs JE, Martin-Marcos P, Saini AK, Hinnebusch AG, Lorsch JR. 2009. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J Mol Biol 394:268–285. doi: 10.1016/j.jmb.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valásek L, Phan L, Schoenfeld LW, Valásková V, Hinnebusch AG. 2001. Related eIF3 subunits TIF32 and HCR1 interact with an RNA recognition motif in PRT1 required for eIF3 integrity and ribosome binding. EMBO J 20:891–904. doi: 10.1093/emboj/20.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin-Marcos P, Nanda J, Luna RE, Wagner G, Lorsch JR, Hinnebusch AG. 2013. β-Hairpin loop of eukaryotic initiation factor 1 (eIF1) mediates 40 S ribosome binding to regulate initiator tRNA(Met) recruitment and accuracy of AUG selection in vivo. J Biol Chem 288:27546–27562. doi: 10.1074/jbc.M113.498642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin-Marcos P, Cheung YN, Hinnebusch AG. 2011. Functional elements in initiation factors 1, 1A, and 2β discriminate against poor AUG context and non-AUG start codons. Mol Cell Biol 31:4814–4831. doi: 10.1128/MCB.05819-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barth-Baus D, Bhasker CR, Zoll W, Merrick WC. 2013. Influence of translation factor activities on start site selection in six different mRNAs. Translation 1:e24419. doi: 10.4161/trla.24419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 55.Firth AE. 2014. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res 42:12425–12439. doi: 10.1093/nar/gku981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCluskey K, Wiest A, Plamann M. 2010. The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J Biosci 35:119–126. doi: 10.1007/s12038-010-0014-6. [DOI] [PubMed] [Google Scholar]
- 57.Wu C, Wei J, Lin PJ, Tu L, Deutsch C, Johnson AE, Sachs MS. 2012. Arginine changes the conformation of the arginine attenuator peptide relative to the ribosome tunnel. J Mol Biol 416:518–533. doi: 10.1016/j.jmb.2011.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 60.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jacobs GH, Chen A, Stevens SG, Stockwell PA, Black MA, Tate WP, Brown CM. 2009. Transterm: a database to aid the analysis of regulatory sequences in mRNAs. Nucleic Acids Res 37:D72–D76. doi: 10.1093/nar/gkn763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Superimposing conservation of NCCs in cpc-1 homologs on the phylogenetic trees of Pezizomycotina and Agaricomycetes. (A) Phylogeny of Pezizomycotina based on CPC1 amino acid sequences and conservation of NCC6, NCC7, and NCC8. The unrooted phylogenetic tree is based on the amino acid sequences (from the 5′ end to the mORF stop codon when no in-frame stop codon is present or from the stop codon in the 5′ region to the mORF stop codon) of the cpc-1 homologs from the Pezizomycotina species indicated on the right of each branch. Species names are coded with respect to which NCCs are identical to N. crassa: black, NCC6, NCC7, and NCC8; blue, NCC7 and NCC8; green, NCC6 and NCC8; red, NCC8; orange, no identical NCCs. (B) Phylogeny of Agaricomycetes based on CPC1 amino acid sequences and conservation of a single NCC. The unrooted phylogenetic tree is based on the amino acid sequences (from the 5′ end to the mORF stop codon or from the stop codon in the 5′ region to the mORF stop codon) of the cpc-1 homologs from the Agaricomycetes species indicated on the right of each branch. The asterisk indicates a sequence presumed to derive from an unknown Agaricomycetes species contaminating the Pinus taeda EST library. Species names in black, NCC = AUU; names in blue, NCC = UUG; in orange, NCC = AUA; in red, NCC = ACG or CUG. The trees were generated using ClustalX. Bootstrap values are given for key nodes in Pezizomycotina. The scale represents the divergence rate at each residue. Download FIG S1, PDF file, 0.2 MB (160.5KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Conservation of NCCs in cpc-1 homologs of different fungi. (A and B) Nucleotide sequences used for generating WebLogo in Fig. 1 and S5, respectively. Species identifiers of the sequences used are indicated on the left. An asterisk next to the species name indicates sequence presumed to derive from unknown Pezizomycotina or Agaricomycetes that is contaminating an EST library of the indicated species (e.g., Agaricomycetes sequence contaminating Pinus taeda library). Presumed start codons are highlighted in green. Sequences from species names in quotation marks are presumed contaminants of fungal origin because they cluster with cpc-1 from Pezizomycotina or Agaricomycetes even though the name of the sequenced species is a plant. Alignments of the 10 nucleotides including the AUG or NCC start codons and the nucleotides that define the initiation contexts (−6 to +4) which were used to generate the WebLogo in Fig. 1C to J and S5D and E are shown for cpc-1/GCN4 homologs from uAUG1 uAUG2, mAUG, NCC6, NCC7, and NCC8 from Pezizomycotina (A) and the single NCC from Agaricomycetes (B). (C) Nucleotide sequences of the 5′ regions of cpc-1 homologs from the upstream in-frame stop codon to the uAUG in Tuber melanosporum and Arthrobotrys oligospora. NCCs are highlighted in green. Download FIG S2, PDF file, 0.3 MB (265KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Regions upstream of mAUG show coding conservation in frame with the mORF. (A) Conservation in the N-terminal extension of N. crassa cpc-1. (i and ii) Nucleotide and codon alignments of the region corresponding to uORF2 of cpc-1 in N. crassa are shown in frame with the main ORF (frame 1 in Fig. 1B) (i) and in frame with uORF2 (frame 3 in Fig. 1B) (ii). (iii to v) A proline-rich region (residues 218 to 224, red dashed bracket in Fig. 1K) upstream of the cpc-1 mAUG shows high conservation in the mORF. Nucleotide and codon alignments of this region are shown for frame 1 (in frame with the mORF) (iii), frame 2 (iv), and frame 3 (v). In these alignments, consensus codons (and gaps) are highlighted in white, synonymous codons in green, nonsynonymous codons in brown, and stop codons in magenta. Species identifiers of the sequences used are indicated on the left. The numbers of synonymous and nonsynonymous substitutions are summed below each panel. The additional numbers in parentheses below panel i indicate the number of synonymous and nonsynonymous substitutions in frame with the mORF in the small patch of sequence marked by a dashed orange bracket above the aligned sequences. This patch represents the conserved peptide region indicated by the corresponding dashed orange bracket in Fig. 1K. The alignments were generated by a custom script that conceptually translated the main ORF frame into peptide sequence, then performed ClustalX alignment of peptide sequence, and then converted this alignment back to nucleotides. The most common codon in each column was then designated the consensus codon; synonymous and nonsynonymous substitutions were determined relative to it. (C) The region corresponding to the beginning of uORF3 of Coprinus cinerea cpc-1 is conserved at the amino acid level in frame with the mORF in Agaricomycetes homologs. Nucleotide and codon alignments of the region, including the beginning of uORF3 of C. cinerea cpc-1 (indicated by red dashed lines in Fig. S5F), are shown for frame 1 (in frame with the main ORF) (vi), frame 2 (in frame with uORF3) (vii), and frame 3 (viii). Species identifiers of the sequences used are indicated on the left. “Pinus_taeda_cont” indicates a sequence coming from an unknown Agaricomycetes species contaminating the Pinus taeda EST library. The numbers of synonymous and nonsynonymous substitutions are indicated on the right of each panel. The position of the AUG start codon of uORF3 is indicated by an inverted red triangle. Coloring scheme, alignment generation, and display are the same as in other panels. Download FIG S3, PDF file, 2.6 MB (2.7MB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Coding potential and synonymous site conservation in cpc-1. At the top is a map of the N. crassa maximal CPC1-frame ORF region, showing the main CPC1 ORF; uNCC6, uNCC7, and uNCC8; and uORFs 1 and 2. Immediately below is shown the coding potential in the three reading frames as measured with MLOGD using an alignment of the maximal CPC1-frame ORF regions from 96 fungal species and a 20-codon sliding window. MLOGD uses a principle similar to the dN/dS statistic (ratio of nonsynonymous to synonymous substitutions) but also accounts for conservative amino acid substitutions being more probable than nonconservative substitutions. Positive scores indicate that the sequence is likely to be coding in the given reading frame and are observed throughout CPC1 and most of the upstream extension, at least from uNCC8. Positions where >50% of the alignment divergence is missing due to alignment gaps are omitted. Below this is shown alignment divergence (aln. divg, mean number of substitutions per nucleotide) for the sequences that contribute to the statistics at each position in the alignment (in any particular column, some sequences may be omitted from the statistical calculations due to alignment gaps). The bottom panels show the analysis of synonymous site conservation across the alignment. The brown line (“o/e”) indicates the ratio of the observed number of synonymous substitutions within a 5-codon window to the number expected under a null model of neutral evolution at synonymous sites, while the red line (“p-val”) depicts the corresponding statistical significance. Download FIG S4, PDF file, 0.1 MB (138.1KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Features of cpc-1 homologs from Basidiomycota. (A to C) Diagrams of the cpc-1 transcripts from different Basidiomycota branches. AUG codons (green bars), stop codons (red bars), mORFs (black bars), and uORFs (blue bars) are indicated. The numbers of sequences used for the comparisons are shown in parentheses. Features are drawn to scale. (A) Ustilago maydis (representing the three available Ustilaginomycotina sequences). Conserved NCCs in frame 1 upstream of uORF2 are indicated by magenta bars. (B) Coprinopsis cinerea (representing the 27 available Agaricomycetes sequences containing uORFs). The position of the single conserved NCC in frame 1 upstream of uORF3 is indicated by a magenta bar. (C) Leucosporidium scottii (representing the two available Microbotryomycetes sequences containing uORFs). The position of the single conserved NCC in frame 1 upstream of the uORF is indicated by a magenta bar. (D and E) Frequency WebLogo of the conservation of the initiation contexts, from −6 to +4, of all C. cinerea genes (D) and the single conserved NCC in frame 1 of Agaricomycetes upstream of the uORFs (E). (F) The amino acid sequence encoded by frame 1 of C. cinerea cpc-1 mRNA (the mORF), bounded by two in-frame stop codons. mAUG indicates the annotated initiation codon; the position of the conserved upstream NCC is also indicated. The approximate positions of uORF1, uORF2, and uORF3 are indicated by blue lines. The C-terminal bZIP domain is indicated in red font. The region analyzed in Fig. S3C is bracketed by a dashed red line. The conservation of each residue from the alignment of homologs from 27 species (those used to construct the tree in Fig. S1B) is shown below the amino acid sequence. Download FIG S5, PDF file, 0.3 MB (311KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The sequences of cpc-1–luc fusion constructs used for in vitro experiments. Mutations and selected unique restriction sites are shown below the sequence. uORF1, uORF2, and the beginning of the main ORF (mORF) are shaded gray. NCCs are shaded magenta and are numbered 1 to 8. The position of the introduced TAA mutation that terminates the translation from NCCs is indicated by a red box. Luciferase was placed in frame with CPC1 at the XhoI site. Download FIG S6, PDF file, 0.2 MB (217.6KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Toeprint analysis indicates reinitiation following translation of cpc-1 uORF1 but not uORF2 (primary data for Fig. 2B). The description of the experiment is given in the text. For markers, either primer CPC101 (upper panel) or ZW4 (lower panel) was used to sequence the WT template (the four leftmost lanes). The nucleotide complementary to the dideoxynucleotide added to each sequencing reaction for the wild-type cpc-1–luc template is indicated above the corresponding lane so that the sequence of the template can be directly deduced; the 5′-to-3′ sequence reads from top to bottom. Asterisks (top to bottom), toeprint products corresponding to ribosomes bound at uAUG1, uAUG2, and mAUG on the cpc-1–luc RNA. Boxes (top to bottom), uAUG1, uAUG2, and mAUG in the sequence markers. The ribosome protects approximately 16 nt 3′ of a start codon in the ribosome P-site. Download FIG S7, PDF file, 0.3 MB (287.4KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Evidence from [35S]Met labeling showing that NCC5 to -8, but not NCC1 to -4, initiate translation in an N. crassa cell-free system. Synthetic RNAs (60 ng) for the indicated constructs were used to program 10 µl of cell-free translation reaction mixtures from N. crassa. Reaction mixtures were incubated for 30 min at 26°C. Radiolabeled products were analyzed on 12% NuPAGE gels. Open circles, translation products eliminated upon mutation of NCC5 to -8; the product predicted to be initiated from NCC8 also increased when NCC8 was changed to AUG (lane 12). Arrowhead, position of mAUG-initiated translation product (mORF). Brackets, translation products larger than the mORF produced in the absence of an in-frame UAA stop codon. Download FIG S8, PDF file, 0.1 MB (134KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ribosome profiling data, including framing information, for arg-2 and eif5. The ribosome-protected fragment count for each position 5′ to 3′ along the mRNA is shown on top for arg-2 (NCU07732) (A) and eif5 (NCU00366) (B). Peaks corresponding to specific NCCs in the 5′ leader of eif5 are shown by red arrows. A schematic map of the ORF organization, drawn to scale, is shown below the ribosome-protected fragment count graph. The reading frame information derived from the ribosome-protected fragments for specific regions along the mRNAs is shown below the schematic maps. AAP, uORF-encoded arginine attenuator peptide; all CDSs, all predicted N. crassa genic ORFs. The color coding for different components of the figure is the same as in Fig. 5. Download FIG S9, PDF file, 0.2 MB (252.8KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmids (A) and primers (B) used in the study. Download TABLE S1, PDF file, 0.1 MB (101.9KB, pdf) .
Copyright © 2017 Ivanov et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.