

Abstract
Background
Continuing evolution of the Mycobacterium tuberculosis (Mtb) complex genomes associated with resistance to anti-tuberculosis drugs is threatening tuberculosis disease control efforts. Both multi- and extensively drug resistant Mtb (MDR and XDR, respectively) are increasing in prevalence, but the full set of Mtb genes involved are not known. There is a need for increased sensitivity of genome-wide approaches in order to elucidate the genetic basis of anti-microbial drug resistance and gain a more detailed understanding of Mtb genome evolution in a context of widespread antimicrobial therapy. Population structure within the Mtb complex, due to clonal expansion, lack of lateral gene transfer and low levels of recombination between lineages, may be reducing statistical power to detect drug resistance associated variants.
Results
To investigate the effect of lineage-specific effects on the identification of drug resistance associations, we applied genome-wide association study (GWAS) and convergence-based (PhyC) methods to multiple drug resistance phenotypes of a global dataset of Mtb lineages 2 and 4, using both lineage-wise and combined approaches. We identify both well-established drug resistance variants and novel associations; uniquely identifying associations for both lineage-specific and -combined GWAS analyses. We report 17 potential novel associations between antimicrobial resistance phenotypes and Mtb genomic variants.
Conclusions
For GWAS, both lineage-specific and -combined analyses are useful, whereas PhyC may perform better in contexts of greater diversity. Unique associations with XDR in lineage-specific analyses provide evidence of diverging evolutionary trajectories between lineages 2 and 4 in response to antimicrobial drug therapy.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5615-3) contains supplementary material, which is available to authorized users.
Keywords: Drug resistance, Evolution, Mutations, Mycobacterium tuberculosis, Tuberculosis
Background
Despite clonal expansion and a lack of lateral gene transfer in Mycobacterium tuberculosis (Mtb), the evolution of drug resistance is threatening tuberculosis disease (TB) control efforts. Resistance to all anti-Mtb drugs has been observed, usually evolving relatively shortly after their introduction. Drug-resistant TB is phenotypically categorised as multi-drug resistant (MDR) when resistant to two first–line drugs, rifampicin and isoniazid; extensively drug-resistant (XDR) occurs when MDR Mtb have additional resistance to fluoroquinolones and at least one second-line injectable. Only 50% of patients receiving treatment for MDR TB, globally, were successfully treated in 2014 [1].
De novo emergence of drug resistance has been observed, with the presence of multiple unfixed drug-resistance mutations and selective sweeps in Mtb populations within patients [2–4]. Additionally, transmission of resistant strains is frequently observed [5, 6]. Indeed, many mutations associated with antimicrobial resistance have been identified [7], some have been associated with no fitness cost and others with additional compensatory mutations that may increase fitness and enable transmission [8]. These polymorphisms include both point mutations, for example, single nucleotide polymorphisms (SNPs) such as in rpoB [9] and structural variants such as the dfrA-thyA double deletion linked to para-aminosalicylic acid resistance [10]. Genes involved in resistance to some drugs are well known; for example, mutations for rifampicin (in rpoB and rpoC) and isoniazid (in katG) are well characterised [7]. However, the mechanisms for ethambutol (embB), pyrazinamide (pncA) and second line drug resistance are not fully known. As whole genome sequencing of Mtb becomes more routinely applied [11], association approaches using genomic variation have the potential to provide new insights into these resistance mechanisms. Compensatory mutations such as those in rpoA and rpoC, associated with the rpoB rifampicin resistance mutations, have been associated with transmission of drug resistant strains [12]. Furthermore, as patients receive a cocktail of anti-Mtb drugs, multiple concomitant resistance can arise naturally, and this complicates the analysis of phenotype-genotype relationships [13].
The genome-wide association study (GWAS) approach has been widely used in human genetics; for example, to identify variants in the class II human leukocyte antigens (HLA) region associated with susceptibility to TB infection [14]. However, it is increasingly being applied to pathogen research and shows great promise [13, 15, 16]. It allows the identification of variants across the genome, associated with specific phenotypes. In order to prevent spurious associations, pathogen GWASs face the need to deal with the much higher levels of population structure seen in bacteria compared to humans, whilst maximising sensitivity [17, 18]. This is especially important for Mtb due to its clonality. This clonality is consistent with a phylogenetic tree structure and thus has led to the application of convergence-based methods, which have identified resistance mutations in Mtb [13, 19]. Such methods seek to identify convergent evolution in genetically diverse strains with similar resistance phenotypes. This happens when mutations in the same gene or nucleotide position occur repeatedly and independently become fixed, thus signaling their positive selection for a particular phenotype.
However, there remain questions as to the importance of historic genetic background variation in the evolution of drug resistance, such as between Mtb lineages, which have not been systematically explored [20]. The Mtb complex is categorised into seven lineages, defined on the basis of molecular typing, which are endemic in different locations around the globe. These lineages are known to have other distinctive features, with some persisting in geographical regions (lineages 5 and 6 in West Africa) and others spreading across continents (lineage 2- East Asian and lineage 4 – Euro-American strains). This observation has led to the hypothesis that the strain-types are specifically adapted to people of different genetic backgrounds [21]. These lineages may vary in their propensity to transmit, their virulence, site of infection and ultimately propensity to cause disease [22–24], but results are inconsistent and there is considerable inter-strain variation within lineages [25, 26]. Recent research into lineage 4 alludes to this variation, suggesting different evolutionary strategies are employed by different sublineages [27]. A set of single nucleotide polymorphisms (SNPs) has been identified that can be used to barcode sub-lineages [28], leading to informatic tools that position sequenced samples within a global phylogeny [29]. Thus, lineage-based genetic differences may also be important in resistance adaptations to anti-Mtb drug exposure.
The current study applies lineage-specific and lineage-combined GWAS, alongside convergence-based PhyC methods, to gain insight into lineage-specific drug resistance evolution. We focus on the modern lineage 2 and lineage 4 isolates, which are known to be drug resistant globally, and use a large dataset involving Mtb isolate sequences from more than 12 countries (n > 4400).
Results
Genomic variants and population structure
High quality SNP and insertion and deletion (indel) variants were characterised in relation to the H37Rv reference genome, from raw sequence data from a convenience sample of existing data for isolates in lineages 2 (n = 702) and 4 (n = 3706). These isolates are within a global drug resistance data set [13], which has been further complemented by additional phenotypic data (see Methods). After removing variants that are monomorphic within each dataset, the final lineage-combined dataset consisted of 157,726 SNPs, 5998 deletions and 2926 insertions across the 4408 isolates (see Additional file 1). The median number of SNPs per sample in the lineage 2 dataset, after removing monomorphic variants, was 332 (range: 189–386) and in lineage 4 was 724 (range: 10–870) (significant difference between lineages with Wilcoxon test p-value < minimum calculable (2.2 × 10− 16)). Lineage 4 contains the H37Rv reference strain, but also has increased strain-type diversity [13, 28]. The median number of indels per sample in lineage 2 was 31 (range: 7–42) and in lineage 4 was 40 (range: 2–61) (significant difference between lineages Wilcoxon test: p-value < minimum calculable (2.2 × 10− 16)) (see Additional file 1). The majority of variants were rare, with 75% of them found to have a non-reference variant frequency (defined as the number of isolates with a non-reference allele at a specific variant position divided by the total number of isolates with a non-missing allele at this position) of less than 0.0028 and 0.00054 in lineages 2 and 4, respectively (see Additional file 1 and Additional file 2). A principal component analysis (PCA) using the variants revealed the expected clustering by lineage and greater diversity within lineage 4 (see Additional file 3). Within lineage 2, the first 10 principal components account for 71.9% of the variation (see Additional file 3 and Additional file 4) and the mean pairwise variant distance was 1074 (range: 0–6270) (see Additional file 3). Within lineage 4, the first 10 principal components account for 88.9% of the variation (see Additional file 3 and Additional file 4) and the mean pairwise variant distance was 1458 (range: 0–11,780) (see Additional file 3). There are 567 isolates with < 10 variants different from at least one other isolate, indicative of potential transmission events, which can confound an association analysis. A phylogenetic tree constructed using the variants mimicked the relationships observed in the PCA, with isolates clustering by sublineage on both (see Additional file 3 and Fig. 1).
Fig. 1.

Phylogenetic tree of all samples: coloured by phenotype, sublineage and genotype for novel variants alongside katG, rpoB and gyrA. From inner to outer, each track is coloured by; drug-resistance phenotype, sublineage, variant genotype for; hadA (732110), Rv3115-moeB2 (3482717), whiB6-Rv3863 (4338594), Rv0197 (232574), recF (4047), argJ (1867614), katG (2155168); locus genotype for; Rv1313c-Rv1314c, fadB4-Rv3142c, Rv0526, espE-espF, tuf, cut5b-Rv3725, Rv3007c, Rv2668, pip-Rv0841, moeX, lipJ-cinA, Rv3128c-Rv3129, rpoB, gyrA. Variant genotype is coloured in samples where a non-reference variant is present with respect to H37rv reference and variant position is given in brackets. Locus genotype is coloured in samples with one or more non-synonymous or intergenic variants at each locus with respect to H37rv reference, relative height of the bar reflects number of variants at each loci
Drug resistance phenotypes
Overall, analyses were conducted for 17 drug resistance phenotypes, including for 12 individual drugs and 5 composite phenotypes. The 12 individual drug resistance phenotypes with frequency of resistance ranging from 3.3% (MOX in lineage 4) to 43.0% (STM in lineage 2), and the composite phenotypes of MDR (lineage 2 35.7%; lineage 4 9.5%) and XDR (lineage 2 9.9%; lineage 4 1.2%). The combined second-line drug resistance phenotypes for resistance to any fluoroquinones (FQ) and resistance to any aminoglycosides (AG) were also considered (see Additional file 5). The completeness of drug-resistance phenotype data is variable. Rifampicin was the most tested for (tested for in 92.0% of isolates); while ciprofloxacin was the least (tested for in 4.2% of isolates) (see Additional file 6). Furthermore, there is evidence of multiple concomitant resistance with 44.1% of MDR isolates also resistant to ethambutol.
Convergence-based analyses, variant-based GWAS and locus-based identified known resistance conferring variants
We performed convergence-based analyses (PhyC), GWAS across loci (locus-based) and GWAS on individual variants (variant-based). Each were conducted in a lineage-specific and lineage-combined manner. Due to the close relatedness between some samples, for the GWAS analyses, we applied specialized regression models with random effects that have been implemented in a human setting to handle “cryptic relatedness” [13] (see Methods).
In total, PhyC analysis of the combined lineages identified 53 variants in 20 different loci, with individual lineage analyses identifying a subset of these loci (see Table 1, Additional file 7). Eleven of these loci were not identified by GWAS techniques, including eight loci with known involvement in antimicrobial resistance; thyX-hsdS.1 (para-aminosalicylic acid), rpoC (rifampicin), pncA-Rv2044c (pyrazinamide), eis-Rv2417c (aminoglycosides), folC (para-aminosalicylic acid), fabG1 (isoniazid), oxyR’-ahpC (isoniazid) and gyrB (fluoroquinolones) (see Table 1, Additional file 8).
Table 1.
Significant associations between genomic variants and drug resistance phenotypes identified by PhyC
| Locus | Known Phenotype Association [13] | Lineage | Observed Phenotype Association (position; p-value) | Total |
|---|---|---|---|---|
| rpoB | RMP | 2 | INH(761,155;1.2 × 10− 06, 761,139;5.6 × 10− 06), MDR(761,155;1.5 × 10− 12, 761,139;1.4 × 10− 07, 761,140;5.5 × 10− 04), RMP(761,155;2.4 × 10− 11, 761,139;6.7 × 10− 09, 761,110;4.6 × 10− 04, 761,140;9.3 × 10− 04), XDR(761,155;2.6 × 10− 04, 761,139;7.5 × 10− 04) | 11 |
| rpoB | RMP | 4 | AG(761,155;2.0 × 10−5), EMB(761,155;2.2 × 10− 14), INH(761,155;3.4 × 10− 32, 761,139;6.2 × 10− 11, 761,110;7.6 × 10− 08, 761,140;3.3 × 10− 07), MDR(761,155;2.1 × 10− 45, 761,139;2.8 × 10− 20, 761,140;1.3 × 10− 09, 761,110;1 × 10− 08, 759,939;2.8 × 10− 04), PZA(761,155;1.5 × 10− 12, 761,110;2.2 × 10− 06), RMP(761,155;5.7 × 10− 54, 761,139;9.7 × 10− 27, 761,110;2.6 × 10− 12, 761,140;3.4 × 10− 11, 761,998;1.1 × 10− 05, 761,109;7.3 × 10− 05, 759,939;4.9 × 10− 04), STRx10P(761,155;2.1 × 10− 16, 761,110;7.5 × 10− 04), XDR(761,155;7.1 × 10− 14, 761,110;2.7 × 10− 05) | 23 |
| rpoB | RMP | 2 + 4 | AG(761,155;2.3 × 10−5), EMB(761,155;3.8 × 10− 18, 761,140;6.3 × 10− 06, 761,110;1.1 × 10− 05, 761,139;2 × 10− 05), INH(761,155;2.3 × 10− 42, 761,139;3.5 × 10− 19, 761,140;2.3 × 10− 11, 761,110;1.1 × 10− 09, 761,161;1.5 × 10− 06), MDR(761,155;7.8 × 10− 63, 761,139;1.7 × 10− 31, 761,140;4.8 × 10− 15, 761,110;1.3 × 10− 13, 761,161;5.8 × 10− 05, 761,095;1.8 × 10− 04, 759,939;2.6 × 10− 04, 761,109;2.6 × 10− 04), PZA(761,155;5.6 × 10− 16, 761,110;2 × 10− 07, 761,139;5.8 × 10− 04), RMP(761,155;3.5 × 10− 70, 761,139;3.2 × 10− 38, 761,110;2.4 × 10− 17, 761,140;2.2 × 10− 15, 761,161;4.4 × 10− 07, 761,109;3.5 × 10− 05, 761,998;1 × 10− 04, 761,095;3.1 × 10− 04, 759,939;4.7 × 10− 04, 760,314;4.7 × 10− 04), STM(761,155;8.4 × 10− 21, 761,110;2.1 × 10− 07, 761,139;2.6 × 10− 07, 761,140;8.5 × 10− 05, 761,161;1.8 × 10− 04), XDR(761,155;2.2 × 10− 18, 761,110;2.6 × 10− 08, 761,139;9.7 × 10− 08, 761,161;1.9 × 10− 06, 761,109;6.2 × 10− 05) | 40 |
| embB | EMB | 2 | EMB(4,247,429;3 × 10− 07, 4,247,431;1.8 × 10− 04), INH(4,247,429;9.3 × 10− 10, 4,247,431;3.1 × 10–05), MDR(4,247,429;2.5 × 10− 08, 4,247,431;1.1 × 10− 04), RMP(4,247,429;7.6 × 10− 09, 4,247,431;1.3 × 10− 04, 4,247,730;8.7 × 10− 04), STM(4,247,429;1 × 10− 05, 4,247,431;1.1 × 10− 04), XDR(4,247,429;4.3 × 10− 06, 4,247,431;1.2 × 10− 04, 4,247,730;1.2 × 10− 04) | 14 |
| embB | EMB | 4 | AG(4,247,431;7.1 × 10− 4), EMB(4,247,431;3 × 10− 11, 4,247,729;3 × 10− 08, 4,247,730;1 × 10− 07, 4,248,003;1 × 10− 07, 4,247,429;1.5 × 10− 06, 4,247,574;8.1 × 10− 04), INH(4,247,431;6.6 × 10− 19, 4,247,730;1.4 × 10− 09, 4,247,429;1.7 × 10− 09, 4,247,729;6.3 × 10− 06, 4,247,574;1.8 × 10− 05, 4,248,003;2.8 × 10− 05), MDR(4,247,431;2.3 × 10− 21, 4,247,429;1.5 × 10− 09, 4,247,730;8.8 × 10− 08, 4,247,574;6 × 10− 07, 4,247,729;4.2 × 10− 06, 4,248,003;6 × 10− 04), PZA(4,247,431;1.2 × 10− 04, 4,247,730;2.2 × 10− 04, 4,248,003;5.3 × 10− 04), RMP(4,247,431;2.2 × 10− 18, 4,247,429;1.5 × 10− 10, 4,247,730;1.4 × 10− 08, 4,247,574;6.6 × 10− 05, 4,247,729;1.3 × 10− 04, 4,248,003;1.5 × 10− 04), STM(4,247,431;1.5 × 10− 08, 4,247,729;3.6 × 10− 06, 4,247,574;7.2 × 10–04), XDR(4,247,429;1.3 × 10− 04) | 31 |
| embB | EMB | 2 + 4 | AG(4,247,431;3.5 × 10−4), EMB(4,247,429;9.2 × 10−21, 4,247,431;1.5 × 10− 16, 4,247,729;2.1 × 10− 09, 4,247,730;6.4 × 10− 08, 4,248,003;2.8 × 10− 07, 4,247,574;1.4 × 10− 04, 4,249,518;1.9 × 10− 04), FQ(4,247,730;9.5 × 10− 07), INH(4,247,429;2.7 × 10− 27, 4,247,431;1 × 10− 25, 4,247,730;8.4 × 10− 14, 4,248,003;1.2 × 10− 08, 4,247,729;1.4 × 10− 07, 4,247,574;1.7 × 10− 07), MDR(4,247,431;3.2 × 10− 26, 4,247,429;6.1 × 10− 26, 4,247,730;2 × 10− 12, 4,247,574;2.5 × 10− 09, 4,247,729;1.3 × 10− 07, 4,248,003;1.5 × 10− 07), PZA(4,247,730;6.3 × 10− 08, 4,247,431;2 × 10− 05, 4,247,429;2.9 × 10− 04, 4,248,003;4.6 × 10− 04), RMP(4,247,429;4.1 × 10− 29, 4,247,431;4.8 × 10− 24, 4,247,730;3.1 × 10− 13, 4,248,003;3.5 × 10− 07, 4,247,574;4.7 × 10− 07, 4,247,729;2.5 × 10− 06, 4,247,469;4.7 × 10− 04), STRx10P(4,247,431;2.2 × 10− 14, 4,247,429;2.9 × 10− 13, 4,247,729;1.4 × 10− 05, 4,248,003;2.6 × 10− 05, 4,247,730;5.5 × 10− 05, 4,247,574;6.9 × 10− 05), XDR(4,247,429;4.4 × 10− 13, 4,247,431;8.9 × 10− 10, 4,247,730;2.6 × 10− 08) | 41 |
| katG | INH | 2 | INH(2,155,168;2.7 × 10− 07), MDR(2,155,168;4.5 × 10− 08), RMP(2,155,168;5.7 × 10− 04), STM(2,155,168;8.3 × 10− 04), XDR(2,155,168;4.1 × 10− 09) | 5 |
| katG | INH | 4 | EMB(2,155,168;1.5 × 10− 07), INH(2,155,168;2 × 10− 63, 2,155,167;8.5 × 10− 05), MDR(2,155,168;3 × 10− 58, 2,155,167;2.8 × 10− 04), PZA(2,155,168;1.5 × 10− 09), RMP(2,155,168;2.9 × 10− 29), STRx10P(2,155,168;2.8 × 10− 11), XDR(2,155,168;1.8 × 10− 14) | 9 |
| katG | INH | 2 + 4 | EMB(2,155,168;4.8 × 10− 11), INH(2,155,168;7.1 × 10− 72, 2,155,167;1.1 × 10− 04), MDR(2,155,168;3.3 × 10− 68, 2,155,167;2.6 × 10− 04), PZA(2,155,168;1.7 × 10− 11), RMP(2,155,168;2.5 × 10− 36), STRx10P(2,155,168;3.9 × 10− 18), XDR(2,155,168;3.5 × 10− 28) | 9 |
| rpsL | STM | 2 | INH(781,687;5.9 × 10− 05), MDR(781,687;5.3 × 10− 05), RMP(781,687;4.8 × 10− 04), STM(781,687;4.1 × 10− 08) | 4 |
| rpsL | STM | 4 | AG(781,687;3.8 × 10− 4), INH(781,687;4.3 × 10− 15), MDR(781,687;3.9 × 10− 12), PZA(781,687;6.1 × 10− 06), RMP(781,687;8.3 × 10− 10), STM(781,687;9.6 × 10− 14, 781,822;2.3 × 10− 04) | 6 |
| rpsL | STM | 2 + 4 | AG(781,687;3.8 × 10− 5), EMB(781,687;3.5 × 10− 05), FQ(781,687;8.3 × 10− 05), INH(781,687;2.3 × 10− 26, 781,822;6.4 × 10− 05), MDR(781,687;2.3 × 10− 25, 781,822;4.1 × 10− 06), PZA(781,687;1.5 × 10− 08), RMP(781,687;4.8 × 10− 22, 781,822;8.6 × 10− 06), STM(781,687;3.4 × 10− 30, 781,822;2.6 × 10− 07), XDR(781,687;4.3 × 10− 09) | 13 |
| Rv1482c-fabG1 | INH, ETH | 2 | INH(1,673,425;9 × 10− 06), MDR(1,673,425;5.7 × 10− 05) | 2 |
| Rv1482c-fabG1 | INH, ETH | 4 | INH(1,673,425;2.2 × 10− 20), MDR(1,673,425;2 × 10− 07), XDR(1,673,425;3.9 × 10− 05) | 3 |
| Rv1482c-fabG1 | INH, ETH | 2 + 4 | EMB(1,673,432;5.4 × 10− 05), ETH(1,673,425;7.6 × 10− 04), FQ(1,673,432;9 × 10− 04), INH(1,673,425;6.4 × 10− 27, 1,673,432;8.3 × 10− 07), MDR(1,673,425;4.4 × 10− 14, 1,673,432;6.4 × 10− 05), RMP(1,673,432;7.9 × 10− 06, 1,673,425;3 × 10− 05), STEP(1,673,432;8.5 × 10− 05, 1,673,425;3.6 × 10− 04), XDR(1,673,425;8.7 × 10− 06, 1,673,432;1.2 × 10− 05) | 14 |
| gyrA | FQ | 2 | EMB(7582;1.3 × 10− 05), ETH(7582;9.5 × 10− 04), FQ(7582;2.3 × 10− 09, 7570;7.6 × 10–06, 7581;4.1 × 10− 04), INH(7582;9.9 × 10− 06), MDR(7582;7.9 × 10− 05), OFL(7582;1.4 × 10− 06, 7570;8.5 × 10− 04, 7581;8.5 × 10− 04), RMP(7582;2.6 × 10− 06, 7570;1.2 × 10− 04, 7581;9.3 × 10− 04), STM(7582;6.5 × 10− 04), XDRvMDR(7570;9.6 × 10− 04), XDR(7570;5.7 × 10− 07, 7582;7.5 × 10− 07, 7581;7.5 × 10− 04) | 21 |
| gyrA | FQ | 4 | EMB(7570;1.2 × 10− 05), FQ(7570;1.9 × 10− 08, 7582;3 × 10− 06, 7581;2.1 × 10− 05), INH(7570;3.2 × 10− 10, 7581;9.2 × 10− 05, 7582;1.1 × 10− 04), KAN(7570;1.5 × 10− 04), MDR(7570;1 × 10− 08, 7582;4.2 × 10− 06, 7581;5 × 10− 05), OFL(7570;2.5 × 10− 04, 7582;5.6 × 10− 04), PZA(7570;1 × 10− 05, 7581;1.3 × 10− 04), RMP(7570;3.4 × 10− 11, 7582;5 × 10− 08, 7581;5.1 × 10− 06), XDR(7570;3.3 × 10− 10, 7582;2.7 × 10− 05, 7572;3.6 × 10− 04) | 24 |
| gyrA | FQ | 2 + 4 | AMK(7570;6.5 × 10− 04), CAP(7570;9.9 × 10− 04), EMB(7582;4.2 × 10− 13, 7570;1.1 × 10− 08, 7572;3.7 × 10− 04, 7581;4.8 × 10− 04), ETH(7582;1.3 × 10− 04), FQ(7582;3.6 × 10− 18, 7570;4.1 × 10− 14, 7581;2.4 × 10− 09, 7572;6.1 × 10− 04), INH(7582;4.5 × 10− 15, 7570;4.2 × 10− 14, 7581;1.2 × 10− 08, 7572;8.3 × 10− 06), KAN(7570;5.7 × 10− 06, 7572;6.7 × 10− 05), MDR(7582;2.2 × 10− 15, 7570;1.8 × 10− 11, 7581;6.4 × 10− 07), OFL(7582;1.3 × 10–10, 7570;2 × 10–07, 7581;5.8 × 10− 06), PZA(7581;1.2 × 10− 07, 7570;2.4 × 10− 07, 7572;9.7 × 10− 05, 7582;4.6 × 10− 04), RMP(7582;2.3 × 10− 20, 7570;2.5 × 10− 17, 7581;1.8 × 10–10, 7572;1 × 10− 06), STM(7582;1.9 × 10− 10, 7570;1.5 × 10− 06, 7581;1.8 × 10− 04), XDRvMDR(7570;4.2 × 10− 05, 7582;5.7 × 10− 04), XDR(7570;3.4 × 10− 19, 7582;3 × 10− 16, 7572;9.7 × 10− 08, 7581;2.9 × 10− 07) | 44 |
| rrs | STM, AG | 2 | AMK(1,473,246;1.8 × 10− 04), CAP(1,473,246;5 × 10− 08), INH(1,473,246;5 × 10− 06), KAN(1,473,246;1.3 × 10− 11), RMP(1,473,246;8.9 × 10− 06), STM(1,473,246;4.1 × 10− 04), XDRvMDR(1,473,246;3.6 × 10− 05), XDR(1,473,246;7.4 × 10− 11) | 8 |
| rrs | STM, AG | 4 | AG(1,473,246;2.6 × 10−7), AMK(1,473,246;4.6 × 10− 06), CAP(1,473,246;2 × 10− 06), CIP(1,473,246;9.4 × 10− 04), EMB(1,473,246;7.1 × 10− 07), FQ(1,473,246;2.5 × 10− 04), INH(1,473,246;3.2 × 10− 10), KAN(1,473,246;3.3 × 10− 10), MDR(1,473,246;4.6 × 10− 06), PZA(1,473,246;9.4 × 10− 10), RMP(1,473,246;1.9 × 10− 16), STM(1,473,246;3.1 × 10− 05, 1,472,359;2.3 × 10− 04), XDRvMDR(1,473,246;1.9 × 10− 05), XDR(1,473,246;1.6 × 10− 13) | 15 |
| rrs | STM, AG | 2 + 4 | AG(1,473,246;7.5 × 10−5), AMK(1,473,246;3.9 × 10− 11), CAP(1,473,246;7.2 × 10− 14), CIP(1,473,246;6.5 × 10− 04), EMB(1,473,246;2.5 × 10− 11), FQ(1,473,246;3.5 × 10− 07), INH(1,473,246;3.6 × 10− 20, 1,472,359;1.2 × 10− 05), KAN(1,473,246;7.9 × 10− 22), MDR(1,473,246;1.8 × 10− 11, 1,472,359;4.4 × 10− 04), PZA(1,473,246;2.6 × 10− 10), RMP(1,473,246;7.3 × 10− 26), STM(1,473,246;1.3 × 10− 11, 1,472,359;1.5 × 10− 08), XDRvMDR(1,473,246;2.1 × 10− 09), XDR(1,473,246;7.9 × 10− 29, 1,472,359;1.5 × 10− 04) | 18 |
| thyX-hsdS.1 | PAS | 2 | XDR(3,067,961;7.5 × 10−04) | 1 |
| thyX-hsdS.1 | PAS | 4 | INH(3,067,961;4.9 × 10−04), STM(3,067,961;3.2 × 10− 04) | 2 |
| thyX-hsdS.1 | PAS | 2 + 4 | EMB(3,067,961;1 × 10− 05), INH(3,067,961;1.4 × 10− 07), MDR(3,067,961;6.4 × 10− 05), RMP(3,067,961;9.4 × 10− 05), STM(3,067,961;2.3 × 10− 07), XDR(3,067,961;1.2 × 10− 05) | 6 |
| rpoC | RMP | 4 | EMB(764,817;2 × 10− 04), MDR(764,817;6 × 10− 07, 764,840;2.8 × 10− 04), PZA(764,817;8.3 × 10− 08), RMP(764,817;3.6 × 10− 08, 764,840;4.9 × 10− 04, 767,123;4.9 × 10− 04), STM(764,817;3.4 × 10− 04) | 8 |
| rpoC | RMP | 2 + 4 | EMB(764,817;1.9 × 10− 04), INH(764,817;1.1 × 10− 05, 764,840;1.1 × 10− 04), MDR(764,817;4.9 × 10− 10, 764,840;9.7 × 10− 06), PZA(764,817;2 × 10− 07), RMP(764,817;1.4 × 10− 09, 764,840;2.2 × 10− 05, 764,363;4.7 × 10− 04, 767,123;4.7 × 10− 04), STM(764,817;3.8 × 10− 06) | 11 |
| embC-embA | EMB | 2 | EMB(4,243,217;1.7 × 10−04) | 1 |
| embC-embA | EMB | 4 | EMB(4,243,221;3.7 × 10−04, 4,243,190;3.8 × 10–04), INH(4,243,221;8.5 × 10− 05), MDR(4,243,217;4.6 × 10− 06, 4,243,221;4.6 × 10− 06, 4,243,190;3.6 × 10− 05), RMP(4,243,221;1.1 × 10− 05, 4,243,190;4.9 × 10− 04) | 8 |
| embC-embA | EMB | 2 + 4 | EMB(4,243,217;1.4 × 10−07, 4,243,190;3 × 10− 07, 4,243,221;1.3 × 10− 06), INH(4,243,217;4 × 10− 08, 4,243,221;2.3 × 10− 06, 4,243,190;3 × 10− 05), MDR(4,243,217;1.2 × 10− 09, 4,243,221;6.9 × 10− 08, 4,243,190;1.9 × 10− 06), RMP(4,243,221;2.2 × 10− 07, 4,243,217;6.1 × 10− 07, 4,243,190;4.8 × 10− 06), STM(4,243,217;8.6 × 10− 04) | 13 |
| hadA | Novel | 4 | INH(732,110;4 × 10−04), MDR(732,110;2.8 × 10− 04) | 2 |
| hadA | Novel | 2 + 4 | INH(732,110;1.1 × 10−04), MDR(732,110;2.6 × 10− 04), STM(732,110;4 × 10− 04) | 3 |
| pncA | PZA | 4 | EMB(2,288,868;3.8 × 10− 04), MDR(2,288,764;2.8 × 10− 04), RMP(2,288,764;4.9 × 10− 04) | 3 |
| pncA | PZA | 2 + 4 | EMB(2,288,820;1.9 × 10− 04, 2,289,103;1.9 × 10− 04), MDR(2,289,207;2.6 × 10− 04), PZA(2,289,207;9.7 × 10− 05), RMP(2,288,778;4.7 × 10− 04, 2,288,820;4.7 × 10− 04) | 6 |
| pncA-Rv2044c | PZA | 4 | RMP(2,289,252;4.9 × 10− 04), XDR(2,289,252;3.6 × 10− 04) | 2 |
| pncA-Rv2044c | PZA | 2 + 4 | INH(2,289,252;1.1 × 10− 04), MDR(2,289,252;5 × 10− 05), PZA(2,289,252;2 × 10− 07), RMP(2,289,252;4.8 × 10− 06), XDR(2,289,252;6.2 × 10− 05) | 5 |
| Rv3115-moeB2 | Novel | 2 + 4 | MDR(3,482,717;6.7 × 10− 04), STM(3,482,717;6.7 × 10− 04) | 2 |
| eis-Rv2417c | AG | 2 + 4 | EMB(2,715,342;1.6 × 10− 05), FQ(2,715,342;1.7 × 10− 04), INH(2,715,342;1.1 × 10− 04), KAN(2,715,342;5.4 × 10− 04), RMP(2,715,342;2.2 × 10− 05), STM(2,715,342;1.4 × 10− 05) | 7 |
| folC | PAS | 4 | EMB(2,747,471;3.8 × 10−04) | 1 |
| folC | PAS | 2 + 4 | EMB(2,747,471;3.7 × 10−04), INH(2,747,471;1.1 × 10− 04), STM(2,747,471;4 × 10− 04) | 3 |
| whiB6-Rv3863 | Putative STM or ETH | 2 + 4 | EMB(4,338,594;9 × 10− 04) | 1 |
| fabG1 | INH [53] | 4 | INH(1,674,048;6.3 × 10− 06) | 1 |
| fabG1 | INH [53] | 2 + 4 | INH(1,674,048;5.5 × 10− 06) | 1 |
| oxyR’-ahpC | INH | 2 + 4 | INH(2,726,141;6.8 × 10− 04), PZA(2,726,141;9 × 10− 04) | 2 |
| gyrB | FQ | 4 | RMP(6620;4.9 × 10− 04) | 1 |
| gyrB | FQ | 2 + 4 | RMP(6620;4.7 × 10−04) | 1 |
(p-values < 1E-3) Drug resistance phenotype abbreviations are as given in methods. ‘Total’ refers to the total number of significantly associated variants for the locus and lineage in question. AMK = Amikacin-resistance, AG = Aminoglycoside-resistance, CAP = Capreomycin-resistance, CIP = Ciprofloxacin-resistance, EMB = Ethambutol-resistance, ETH = Ethionamide-resistance, FQ = Fluoroquinolone-resistance, INH = Isoniazid-resistance, KAN = Kanamycin-resistance, MDR = Multidrug-resistant, OFL = Ofloxacin-resistance, PAN = pan-susceptible (no known drug-resistance), PZA = Pyrazinamide-resistance, RMP = Rifampicin-resistance, STM = Streptomycin-resistance, XDR = Extensively drug-resistant
Locus-based GWAS identified 23 different loci (see Table 2, Fig. 2, Additional file 7). Fourteen such loci were identified by locus-based GWAS exclusively; of these 14 loci, gid is known to be involved in streptomycin resistance and inhA is known to be involved in isoniazid and ethionamide resistance [30, 31] (see Additional file 8). Variant-based GWAS identified eleven variants in nine different loci. No known associations were identified by variant-based GWAS exclusively; however, three novel associations were identified (RV0197, recF, argJ) (see Table 3, Additional file 8). Three loci were identified by locus-based GWAS and PhyC but not variant-based GWAS: pncA (pyrazinamide), embC-embA and embB (ethambutol) (see Fig. 3a and b, Additional file 8).
Table 2.
Significant associations between loci and drug resistance phenotypes identified by locus-based GWAS
| Locus | Known Phenotype Association [13] | Lineage | Observed Phenotype Association | P-value | PhyC |
|---|---|---|---|---|---|
| rpoB | RMP | 2 | XDR, RMP, MDR | 1.9 × 10− 72, 5.2 × 10− 58, 1.4 × 10− 44 | 11 |
| rpoB | RMP | 4 | RMP, MDR, PZA, XDR, EMB | 2 × 10− 94, 1.1 × 10− 35, 1.1 × 10− 33, 2.5 × 10− 30, 6.4 × 10− 23 | 23 |
| rpoB | RMP | 2 + 4 | RMP, MDR, XDR, PZA | 1.6 × 10− 126, 4.1 × 10− 77, 4.1 × 10− 66, 2.1 × 10− 24 | 40 |
| gyrA | FQ | 2 | MOX, FQ, XDR, OFL | 5.2 × 10− 117, 2.5 × 10− 45, 4.3 × 10− 23, 5.1 × 10− 22 | 21 |
| gyrA | FQ | 4 | FQ, CIP, XDR | 4.1 × 10− 38, 5.4 × 10− 36, 6.5 × 10− 27 | 24 |
| gyrA | FQ | 2 + 4 | FQ, OFL, CIP | 1.2 × 10− 63, 1.7 × 10− 32, 2.2 × 10− 26 | 44 |
| pncA | PZA | 2 | XDR | 1.50 × 10− 25 | 0 |
| pncA | PZA | 4 | PZA | 4.50 × 10− 103 | 3 |
| pncA | PZA | 2 + 4 | PZA, EMB, XDR | 5.3 × 10− 101, 1.2 × 10− 29, 6.7 × 10− 22 | 6 |
| embC-embA | EMB | 4 | XDR, EMB | 6.1 × 10− 69, 2.8 × 10− 26 | 8 |
| embC-embA | EMB | 2 + 4 | XDR, EMB | 3.3 × 10− 37, 1.4 × 10− 23 | 13 |
| katG | INH | 2 | INH, MDR | 3.6 × 10−34, 7.3 × 10−24 | 5 |
| katG | INH | 4 | INH | 1.20 × 10−44 | 9 |
| katG | INH | 2 + 4 | INH, MDR | 1.5 × 10−61, 1.5 × 10−36 | 9 |
| embB | EMB | 2 | EMB | 7.20 × 10−27 | 14 |
| embB | EMB | 4 | EMB | 1.80 × 10−56 | 31 |
| embB | EMB | 2 + 4 | EMB | 3.30 × 10−55 | 41 |
| gid | STM | 4 | STM | 7.40 × 10− 55 | 0 |
| gid | STM | 2 + 4 | STM | 1.30 × 10−53 | 0 |
| Rv1313c-Rv1314c | 4 | XDR | 1.40 × 10−54 | 0 | |
| Rv1313c-Rv1314c | 2 + 4 | XDR | 3.30 × 10−32 | 0 | |
| rpsL | STM | 2 | STM | 1.90 × 10−38 | 4 |
| rpsL | STM | 4 | STM | 5.60 × 10−26 | 6 |
| rpsL | STM | 2 + 4 | STM | 6.00 × 10−41 | 13 |
| fadB4-Rv3142c | 4 | XDR | 4.60 × 10−38 | 0 | |
| Rv0526 | 2 + 4 | XDR | 8.70 × 10−37 | 0 | |
| Rv1482c-fabG1 | INH, ETH | 4 | INH | 1.70 × 10−34 | 3 |
| Rv1482c-fabG1 | INH, ETH | 2 + 4 | INH | 3.30 × 10−30 | 14 |
| espE-espF | 2 + 4 | XDR | 5.70 × 10−31 | 0 | |
| tuf | 4 | XDR | 1.50 × 10−29 | 0 | |
| inhA | INH, ETH | 4 | XDR | 2.40 × 10−28 | 0 |
| cut5b-Rv3725 | 4 | XDR | 5.10 × 10−27 | 0 | |
| Rv3007c | 4 | XDR | 7.80 × 10−24 | 0 | |
| Rv2668 | 4 | XDR | 1.30 × 10−23 | 0 | |
| pip-Rv0841 | 2 + 4 | XDR | 8.60 × 10− 23 | 0 | |
| rrs | STM, AG | 2 | KAN | 1.40 × 10−22 | 8 |
| moeX | 4 | XDR | 5.50 × 10− 22 | 0 | |
| lipJ-cinA | 2 + 4 | XDR | 6.20 × 10−22 | 0 | |
| Rv3128c-Rv3129 | 2 | MDR | 7.40 × 10−22 | 0 |
(P-values <1E-21) Novel associations are given in bold. ‘PhyC’ column refers to the number of associations identified through PhyC analysis for the locus and lineage in question. AMK = Amikacin-resistance, AG = Aminoglycoside-resistance, CAP = Capreomycin-resistance, CIP = Ciprofloxacin-resistance, EMB = Ethambutol-resistance, ETH = Ethionamide-resistance, FQ = Fluoroquinolone-resistance, INH = Isoniazid-resistance, KAN = Kanamycin-resistance, MDR = Multidrug-resistant, OFL = Ofloxacin-resistance, PAN = pan-susceptible (no known drug-resistance), PZA = Pyrazinamide-resistance, RMP = Rifampicin-resistance, STM = Streptomycin-resistance, XDR = Extensively drug-resistant
Fig. 2.

Locus-based GWAS results a Manhattan plot for locus-based GWAS for lineage 2. b Manhattan plot for locus-based GWAS for lineage 4 c Manhattan plot for locus-based GWAS for lineages 2 and 4 combined. P-value threshold <1E-21. Phenotypes with which loci were found to be significantly associated are given in brackets next to locus name. AMK = Amikacin-resistance, AG = Aminoglycoside-resistance, CAP = Capreomycin-resistance, CIP = Ciprofloxacin-resistance, EMB = Ethambutol-resistance, ETH = Ethionamide-resistance, FQ = Fluoroquinolone-resistance, INH = Isoniazid-resistance, KAN = Kanamycin-resistance, MDR = Multidrug-resistant, OFL = Ofloxacin-resistance, PAN = pan-susceptible (no known drug-resistance), PZA = Pyrazinamide-resistance, RMP = Rifampicin-resistance, STM = Streptomycin-resistance, XDR = Extensively drug-resistant
Table 3.
Significant associations between genomic variants and drug resistance phenotypes identified by variant-based GWAS
| Variant Locus | Variant Position | Type | Known Phenotype Association [13] | Lineage | Observed Phenotype Association (p-value) | PhyC |
|---|---|---|---|---|---|---|
| rrs | 1,473,246 | inter | STM, AG | 2 | CAP(2 × 10−31), KAN(1.1 × 10−37) | 8 |
| rrs | 1,473,246 | inter | STM, AG | 4 | KAN(6.7 × 10−31) | 15 |
| rrs | 1,473,246 | inter | STM, AG | 2 + 4 | AMK(2.4 × 10−39), CAP(3.9 × 10−48), KAN(6.5 × 10−69), XDRvMDR(5.3 × 10−27) | 18 |
| katG | 2,155,168 | NS | INH | 2 | XDR(2.1 × 10− 42) | 5 |
| katG | 2,155,168 | NS | INH | 4 | INH(6.1 × 10−65), MDR(6 × 10−45), XDR(1.5 × 10−29) | 9 |
| katG | 2,155,168 | NS | INH | 2 + 4 | INH(4.4 × 10–56), MDR(7.4 × 10−25) | 9 |
| Rv0197 | 232,574 | NS | Novel | 4 | XDR(9.5 × 10−62) | 0 |
| Rv0197 | 232,574 | NS | Novel | 2 + 4 | XDR(232,574;3.8 × 10−51) | 0 |
| rpoB | 761,155 | NS | RMP | 2 | XDR(3.5 × 10− 25) | 4 |
| rpoB | 761,155 | NS | RMP | 4 | MDR(1.2 × 10−27), PZA(1.9 × 10−28), RMP(2.6 × 10− 42, 7.1 × 10−31, 761,139;3.4 × 10−23), XDR(3.8 × 10− 57) | 7 |
| rpoB | 761,139 | NS | RMP | 4 | RMP(3.4 × 10−23) | 3 |
| rpoB | 761,155 | NS | RMP | 2 + 4 | MDR(5 × 10−23), PZA(6 × 10−26), RMP(2 × 10−38), XDR(1.3 × 10− 27) | 7 |
| rpoB | 761,139 | NS | RMP | 2 + 4 | PZA(4 × 10− 23), RMP(2.2 × 10−29), | 7 |
| recF | 4047 | S | Novel | 4 | XDR(1.2 × 10− 52) | 0 |
| recF | 4047 | S | Novel | 2 + 4 | XDR(8.6 × 10−41) | 0 |
| Rv1482c-fabG1 | 1,673,425 | inter | INH, ETH | 4 | INH(1.1 × 10−36) | 3 |
| Rv1482c-fabG1 | 1,673,425 | inter | INH, ETH | 2 + 4 | INH(1.1 × 10− 35) | 14 |
| rpsL | 781,687 | NS | STM | 2 | STM(3 × 10−27) | 4 |
| rpsL | 781,687 | NS | STM | 2 + 4 | STM(6.3 × 10−28) | 6 |
| argJ | 1,867,614 | S | Novel | 2 + 4 | XDR(6.9 × 10−26) | 0 |
| gyrA | 7570 | NS | FQ | 4 | XDR(8.6 × 10−23) | 24 |
| gyrA | 7582 | NS | FQ | 2 + 4 | CIP(1.3 × 10−24), FQ(4.6 × 10−22) | 44 |
(p-values < 1E-22) NS = non-synonymous, S = synonymous, inter = intergenic region. Novel associations are given in bold. ‘PhyC’ column refers to the number of associations identified through PhyC analysis for the locus and lineage in question; AMK = Amikacin-resistance, AG = Aminoglycoside-resistance, CAP = Capreomycin-resistance, CIP = Ciprofloxacin-resistance, EMB = Ethambutol-resistance, ETH = Ethionamide-resistance, FQ = Fluoroquinolone-resistance, INH = Isoniazid-resistance, KAN = Kanamycin-resistance, MDR = Multidrug-resistant, OFL = Ofloxacin-resistance, PAN = pan-susceptible (no known drug-resistance), PZA = Pyrazinamide-resistance, RMP = Rifampicin-resistance, STM = Streptomycin-resistance, XDR = Extensively drug-resistant
Fig. 3.
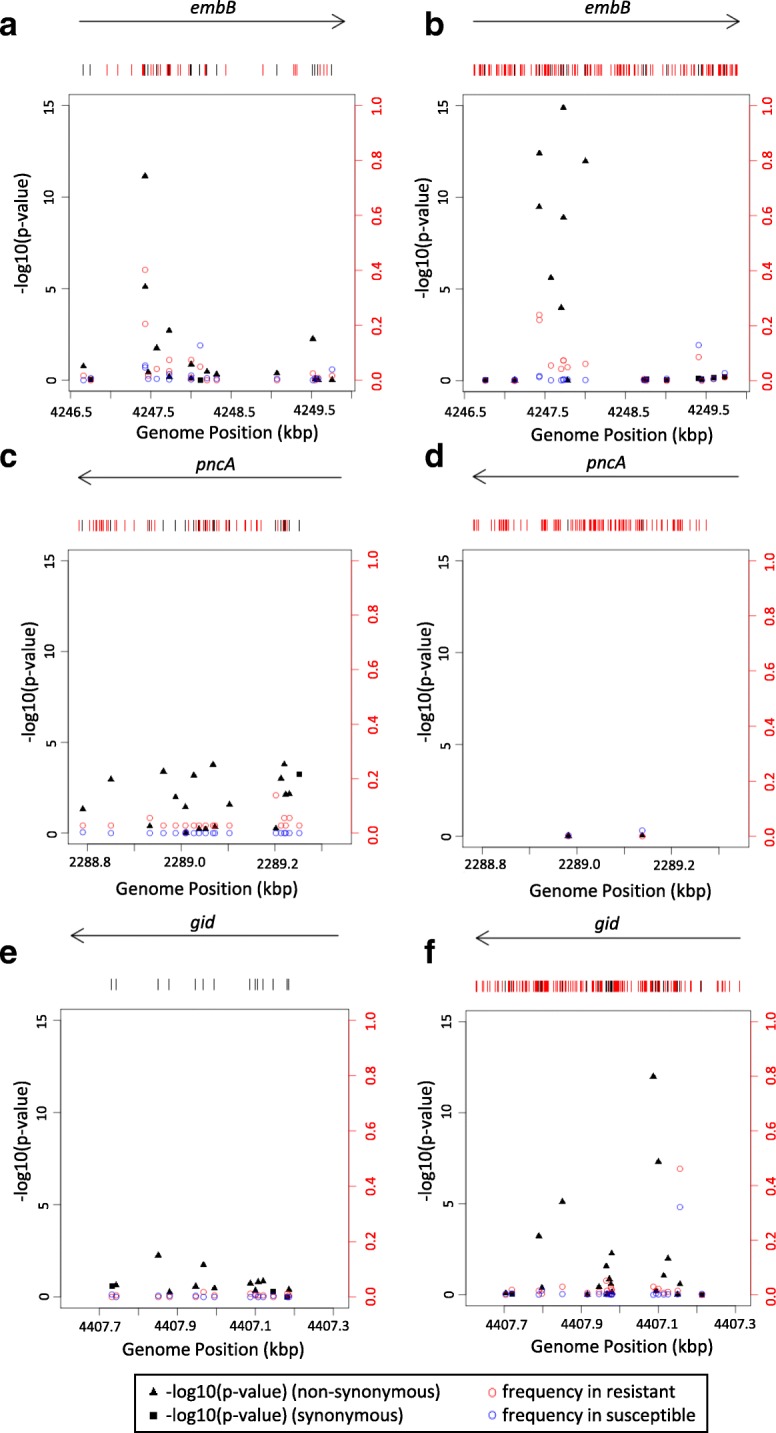
Locus maps showing variant-based GWAS results (on the left y-axis) and variant frequency (on the right y-axis): a lineage 2 ethambutol analysis for embB; b lineage 4 ethambutol analysis for embB; c lineage2 XDR analysis for pncA; d lineage 4 XDR analysis for pncA; e lineage 2 streptomycin analysis for gid; f lineage 4 streptomycin analysis for gid
Effects of lineage-specific analysis on identifying known resistance associated variants
Lineage 2 specific
Overall, for locus-based GWAS analyses across the 16 phenotypes, two loci were identified exclusively to lineage 2 specific analyses; rrs (KAN; p-value = 1.40 × 10− 22) and Rv3128c-Rv3129 (MDR; p-value = 7.4 × 10− 22) (see Fig. 2a). For locus-based GWAS, pncA was found in association with XDR exclusively, however for lineage 4 pncA was found in association with PZA exclusively; greater variation was found in the pncA locus for lineage 2 (see Fig. 3c and d). For the variant-based GWAS analyses there were no lineage 2 exclusive associations. Furthermore, no lineage 2 exclusive associations were identified by PhyC analyses.
Lineage 4 specific
Overall, for the locus-based GWAS analyses, seven loci were identified exclusively by lineage 4 specific analyses (inhA, fadB4-Rv3142c, tuf, cut5b-Rv3725, Rv3007c, Rv2668, moeX) (see Fig. 2b). All of which were found in significant association with the XDR phenotype. For locus-based GWAS, gid was identified in association with streptomycin by lineage 4 specific analyses and –combined analyses but not lineage 2 specific analyses; there is greater variation within the gid locus for lineage 4 (see Fig. 3e and f). The variant-based GWAS analyses identified no lineage 4 exclusive analyses. Moreover, no lineage 4 exclusive associations were identified by PhyC analyses.
Lineages 2 and 4 combined
Four loci were solely identified through combined lineage PhyC analyses; Rv3115-moeB2 (MDR, STM; min. p-value = 6.7 × 10− 4), eis-Rv2417c (STM; min. p-value = 1.4 × 10− 05), whib6-Rv3863 (EMB; p-value = 9 × 10− 4) and oxyR’-ahpC (INH, PZA; p-values = 6.8 × 10− 4, 9 × 10− 4, respectively) (see Table 1). For each loci identified by PhyC, there were consistently the same number or more associations identified by the -combined versus the lineage-specific analyses (see Table 1).
For locus-based GWAS, four loci were identified in association with XDR by the combined lineages 2 and 4 analyses exclusively; Rv0526 (p-value = 8.70 × 10− 37; thioredoxin protein) and three intergenic regions; espE-espF (p-value = 5.70 × 10− 31), pip-Rv0841 (p-value = 8.60 × 10− 23) and lipJ-cinA (p-value = 6.20 × 10− 22) (see Table 2, Fig. 2c).
For variant-based GWAS, one locus was identified by the combined lineages 2 and 4 analyses exclusively; argJ, in association with XDR (p-value = 6.9 × 10− 26) (see Table 3).
Novel resistance-associated variants identified
Across all analyses, we report 17 potentially novel associations between antimicrobial resistance and genomic variants in Mtb; 7 such associations were identified exclusively by lineage-specific analyses (see Tables 1, 2, 3). Twelve were identified by locus-based GWAS, three were identified by variant-based GWAS and two were identified by PhyC. All novel associations identified by GWAS were found in association with the XDR phenotype. There was no overlap in novel associations identified between methods.
Two potentially novel associations were identified by PhyC; hadA (lineage 4, 2 + 4; INH, MDR and STM; 1.1 × 10− 4 < p-values< 4 × 10− 4) and Rv3115-moeB2 (lineages 2 + 4; MDR; STM, min. p-value = 6.7 × 10− 4) (see Table 1, Fig. 1). The Rv3115-moeB2 variant displays a different pattern of variation within lineage 2 than within lineage 4 (see Fig. 1).
Twelve potentially novel associations were identified by locus-based GWAS (see Table 1). Six loci were identified exclusively in lineage 4 all in association with XDR; fadB4-Rv3142c (p-value = 4.6 × 10− 38), tuf (p-value = 1.5 × 10− 29), Rv3007c (p-value = 7.8 × 10− 24), cut5b-Rv3725 (p-value = 5.1 × 10− 27), Rv2668 (p-value = 1.3 × 10− 23) and moeX (p-value = 5.5 × 10− 22). Rv1313c-Rv1314c was identified by both lineage 4 and lineage-combined analyses in association with XDR (min. p-value = 1.4 × 10− 54). Four loci were identified exclusively by lineage-combined analyses in association with XDR; Rv0526 (p-value = 8.7 × 10− 37), espE-espF (p-value = 5.7 × 10− 31), pip-Rv0841 (p-value = 8.6 × 10− 23) and lipJ-cinA (p-value = 6.2 × 10− 22). Rv3128c-Rv3129 was identified exclusively by the lineage 2 analysis in association with MDR (p-value = 7.4 × 10− 22) (see Table 2, Fig. 1).
Three potentially novel associations were identified by variant-based GWAS, all in association with XDR; in the Rv0197 locus (lineage 4, 2 + 4; min. p-value = 9.5 × 10− 62), in the recF locus (lineage 4, 2 + 4; min. p-value = 1.2 × 10− 52, respectively) and the argJ locus (lineages 2 + 4; p-value = 6.9 × 10− 26) (see Table 3, Fig. 1).
Discussion
Our results highlight that lineage specific analyses are able to provide new insights into genetic associations with drug resistance phenotypes, despite a smaller sample size than a pan-lineage approach. Lineage specific associations were found within lineage 2, such as the novel association between Rv3128c-Rv3129 and MDR. We also identified lineage-specific novel associations within lineage 4, such as the association between fadB4-Rv3142c and XDR. This indicates biological differences between these lineages with respect to drug resistance and perhaps in evolutionary trajectory. Novel associations specific to combined analyses indicate convergent evolution between lineages 2 and 4 at the same loci, with variant frequency too low for lineage-specific analyses to detect, that would most likely be detected in larger scale combined analyses (as previously described13). Lineage-specific GWAS is complementary to lineage-combined approaches, with their application in tandem potentially improving the power to detect Mtb genomic variants evolving under differing evolutionary dynamics.
Overall, despite conservative significance thresholds based on permutation, 17 potential novel associations were identified between antimicrobial resistance and Mtb loci and thus warrant experimental validation. For GWAS, 15 novel associations were identified, one in relation to the MDR phenotype and 14 in relation to the XDR phenotype; 7 were lineage specific. This might suggest an evolutionary shift amongst XDR strains. It may be feasible to consider XDR as a highly complex phenotype encompassing transmissibility [32]; unless evolution of XDR from pan-susceptible strains frequently happens within one patient, it is likely that XDR strains have gone through numerous cycles of active disease, transmission and treatment within recent history. The fact that many of these associations are lineage specific lends weight to such a hypothesis, suggesting differing evolutionary trajectories between lineages 2 and 4. Genetic drift might contribute to such divergence; there are numerous bottlenecks during the natural infectious cycle for Mtb, driven by host immune system, anti-TB drug therapy and transmission [33].
Some of the novel associated variants may be involved directly in drug resistance such as hadA, whose gene product, similar to InhA, is involved in fatty acid synthesis type II (FAS-II)) and thus may be involved in isoniazid resistance [34, 35]. One of the novel associated loci, Rv0197, identified here by variant-based GWAS in association with XDR, was previously identified through PhyC in association with a transmissibility phenotype [36]. EspE was identified by this previous analysis also [36], and it remains possible that the espE-espF intergenic region, identified here by locus-based GWAS in association with XDR, may be related by regulation to espE. Additionally, both espE-espF and whiB6-Rv3863 have been linked to Esx-1 which has been implicated in virulence regulation. The WhiB6-Rv3863 intergenic region, which was also identified through previous PhyC analyses including our dataset [13], may additionally be linked to the DosR regulon. This regulon is composed of 48 co-regulated genes and is considered essential for persistence of latent Mtb [37–40]. Interestingly, the whiB6-Rv3863 variant identified shows a markedly different distribution between lineages 2 and 4, showing greater frequency in lineage 2 (see Fig. 1).
Apart from Rv0197, a further two variant-based GWAS SNPs were identified (recF and argJ), however both are synonymous variants. These may be examples of background variants ‘hitchhiking’ alongside causal variants, or may play a biological role. Notably, a number of identified loci are potentially involved in molybdenum cofactor biosynthesis; Rv3115-moeB2, moeX [41], and Rv0197 (mycobrowser: Gene Ontology: molybdenum ion binding) (Mycobrowser). Molybdenum cofactor is found in molybdenum enzymes, which are responsible for a number of functions from dormancy regulation to energy source metabolism [41, 42]. Interestingly, these three loci were each identified by a different analyses type; variant-based GWAS, locus-based GWAS and PhyC, respectively. Functional studies may be useful in providing further insight into the role of variants identified here.
Recognizing that drug resistance phenotypes may be subtly different, depending on the genetic background of the strain, could be important and might relate directly to drug resistance, or to fitness more broadly, such as through increased virulence and transmission. With the recognition of XDR transmission [36, 43], our study suggests that further critical information on lineage and transmission clustering (obtained from the genome sequence) would also be important to determine the full impact of specific mutations, that might lead to further phenotypic descriptions related to transmission, virulence and degree of drug resistance.
The results show the differing evolutionary insights offered by locus- and variant-based GWAS, and convergence-based methodologies. Both variant-based and locus-based GWAS led to unique loci being identified. The rrs locus was found in lineage 2 only locus-based GWAS analyses, but for both variant-based GWAS and PhyC analyses, rrs was identified in both lineage-specific and lineage-combined analyses. Neutral variation within the rrs gene may be diluting the signal from causal drug resistance variants in the lineage 4 locus-based GWAS analysis.
inhA was not identified by variant-based GWAS or PhyC, only lineage 4 specific locus-based GWAS. A sub-type of the Portuguese Lisboa (lineage 4) strain is known to have inhA markers involved in isoniazid resistance [44], and a different mechanism to other lineages. Whilst inhA was not identified by lineage-combined GWAS, it is notable that Rv1482c-fabG1 and katG were; both these loci also play a role in isoniazid resistance, suggesting different mechanisms of resistance to these drugs between lineage 2 and lineage 4.
In cases where drug resistance is driven by rare variants and genetic heterogeneity exists within a single gene, such as in pncA, where multiple alleles can cause pyrazinamide resistance, locus-based analyses may be more powerful. Indeed, pncA was identified here by locus-based GWAS but not variant-based GWAS. Convergence-based PhyC analysis seems to have greater sensitivity in combined-lineage analyses. Unlike GWAS, the success of PhyC in detecting antimicrobial resistance associated variants is determined by the magnitude of convergent evolution within the Mtb population under question [19]. Indeed, there were important differences between the GWAS and PhyC results outlined here. These differences might provide insight into the relative importance of within patient evolution of antimicrobial resistance versus transmission of antimicrobial resistant strains. In instances where a mutation is highly transmissible and consequently increases in frequency with only one or few mutation events, it might be expected that GWAS would be a more powerful analytical tool, due to the lack of convergent-evolution.
It is notable that lineage 2 had a smaller sample size than the lineage 4 dataset, this may contribute to the greater sensitivity in lineage 4 specific analyses. In order to assess the extent to which the lower significance levels in the lineage 2 GWAS were as a result of smaller sample size in comparison to lineage, it would be interesting to repeat the GWAS analyses with a larger and perhaps more geographically spread lineage 2 dataset. Additionally, statistical power is potentially limited in the current analyses by low resolution phenotypic data, with not all drugs tested on all samples, primarily due to second line drugs only being tested where there is multidrug resistance. For example, for lineage 2 there were only 8 resistant and 120 susceptible isolates for moxifloxacin. Despite this, the most significant gene-based GWAS result for lineage 2 was for gyrA, identified in relation to moxifloxacin resistance, showing the sensitivity of the method. Nevertheless, to identify variants with smaller effect sizes, increased phenotypic resolution may prove useful. Further work could explore the use of minimum inhibitory concentration values, where available, being incorporated into resistance phenotypes.
Conclusions
In summary, GWAS and PhyC are sensitive, robust and complementary methodologies in examining evolution of antimicrobial resistance in Mtb. Within GWAS analyses, locus-based and variant-based approaches are both useful and complementary, as are lineage-combined and lineage-specific analyses. These different methodological approaches can be used to detect different evolutionary dynamics and thus their similarities and differences are informative. Evidence presented here suggests the importance of lineage- specific paths of evolution towards drug resistance in Mtb. It will be interesting to see how methodologies outlined here might apply to other Mtb lineages and other pathogen species in an anti-microbial resistance context, or indeed in relation to other phenotypes of interest such as transmissibility.
Methods
Isolates, phenotypic methods, sequencing and variant calling
The raw sequence data used here (n = 4408) form part of a subset of a larger dataset (n = 6465), which represents multiple populations from different geographic areas (see Additional file 9), and is described elsewhere [13]. In particular, only lineages 2 (n = 702) and 4 (n = 3706) from the larger dataset are used, with additional phenotypic data for the samples collected in Portugal. Drug resistance phenotypes were available for amikacin, capreomycin, ciprofloxacin, ethambutol, ethionamide, isoniazid, kanamycin, moxifloxacin, ofloxacin, pyrazinamide, rifampicin, streptomycin, resistance to any fluoroquinolone; levofloxacin, moxifloxacin, ciprofloxacin or ofloxacin (FQ), resistance to any of the aminoglycosides; kanamycin, amikacin, or streptomycin (AG), combined isoniazid and rifampicin resistance, but not XDR (MDR), MDR plus resistance to a fluoroquinolone (ciprofloxacin, levofloxacin, moxifloxacin) and to a second line injectable (amikacin, kanamycin, capreomycin) (XDR), and pan-susceptible, susceptibility to rifampicin and isoniazid plus no other known resistance (PAN). Isoniazid, rifampicin, ethambutol, streptomycin and pyrazinamide are first-line drugs. Amikacin, capreomycin, ofloxacin, para-aminosalicylic acid, moxifloxacin and cycloserine are second-line drugs. Samples found to be MDR, underwent testing for second-line drugs. Para-aminosalycylic acid, levofloxacin, rifabutin and cycloserine resistance phenotypes were excluded from analyses due to lack of data. Where present, levofloxacin data was used in defining the aggregate phenotypes of FQ; however, there was not enough levofloxacin phenotypic data to use in individual drug-resistance analyses.
All samples underwent Illumina sequencing generating paired-end reads of at least 50 bp with at least 50-fold average genome coverage. The raw sequence data were aligned to the H37Rv reference genome (Genbank accession number: NC_000962.3) using the BWA mem algorithm [45]. The SAMtools/BCFtools [46] and GATK [47] software was used to call SNPs and small insertions or deletions (indels) using default options. The overlapping set of variants from the two algorithms was retained for further analysis. Alleles were additionally called across the whole genome (including SNP sites) using a coverage-based approach [16, 28]. A missing call was assigned if the total depth of coverage at a site did not reach a minimum of 20 reads or none of the four nucleotides accounted for at least 75% of the total coverage. The final dataset consisted of 157,726 SNPs, 2926 insertions and 5998 deletions across the 4408 isolates. Monomorphic variants within each of the three datasets (‘lineage 4-specific’, ‘lineage 2-specific’ and ‘lineages 2 and 4 combined’) were removed.
Phylogenetic tree and PhyC
Sublineage was assigned based on SNPs (see Additional file 10). PCA was conducted on the pairwise variant distance matrix for lineages separately and combined. A maximum likelihood phylogenetic tree was constructed for the 157,726 SNP sites present in lineages 2 and 4 isolates using ExaML [48] using the standard model and rooted with M. canettii as the outgroup. The ITOL v3 tool was used for visualisation [49]. PhyC [19] analysis was performed using an in-house pipeline as described by Phelan et al. (2016) [16] . A significance cut-off of < 10− 3 was applied, and this threshold was established based on permutation analysis.
Association analyses
Genome wide association study (GWAS) analyses were performed using GEMMA software [50]. The general parameters were; default missingness (< 0.05) and a minor allele frequency cut-off of 0.001. Kinship matrices were used to account for relatedness. Analyses were performed based on SNPs and short indels (range: 1 to 70 bp) (“variant-based”); and mutations aggregated over coding and intergenic loci (“locus-based”). For coding loci, only non-synonymous variants were aggregated. A linear mixed model was used for both types of analysis, and a likelihood ratio test was used to assess statistical significant of the variants and loci. Each analysis considered a different drug susceptibility phenotype, namely: amikacin resistant (AMK) vs. non-amikacin resistant, AG resistant vs. non-AG resistant, capreomycin resistant (CAP) vs. non-capreomycin resistant, ciprofloxacin resistant (CIP) vs. non-ciprofloxacin resistance, ethambutol resistant (EMB) vs. non-ethambutol resistant, ethionamide resistant (ETH) vs. non-ethionamide resistant, isoniazid resistant (INH) vs. non-isoniazid resistant, kanamycin resistant (KAN) vs. non-kanamycin resistant, moxifloxacin resistant (MOX) vs. non-moxifloxacin resistant, ofloxacin resistant vs. non-ofloxacin resistant (OFL), pyrazinamide resistant (PZA) vs. non-pyrazinamide resistant, rifampicin resistant (RMP) vs. non-rifampicin resistant, streptomycin (STM) vs. non-streptomycin resistant, FQ vs. non-FQ, MDR vs. PAN (“MDR”), XDR vs. PAN (“XDR”) and XDR vs. MDR (“XDRvMDR”). Analyses were performed with lineage 4 only (n = 3706), lineage 2 only (n = 701, after removing 1 outlier identified by PCA) and lineages 2 and 4 combined. Analyses were repeated accounting for different numbers of principal components, from 0 to 5, to assess the effects on significance. A significance threshold of < 10− 21 based on permutation.
All statistical analyses, including PCA, were performed in R software (r-project.org) and its qqman package [51] was used to construct Manhattan plots and quantile-quantile (qq)-plots. Pairwise variant distance between isolates was calculated in R [52], using absolute distance between isolates including all variants for lineage 2 and lineage 4.
Additional files
Variant Summary Tables, Summary tables of variants called in comparison to the H37rv reference, with monomorphic variants removed for each dataset. a Total numbers of variants by lineage; b Number of variants per sample; c Non-reference variant frequency summary; variants called in comparison to the H37rv reference. (PPTX 39 kb)
Non-reference variant frequency histogram, A histogram showing log10(frequency + 1) of non-reference alleles compared to the H37rv reference for a lineage 2 and b lineage4. (PPTX 69 kb)
Population diversity within investigated strains, a Principal component 1 (PC1) by principal component 2 (PC2) for lineage 2, The first 10 principal components account for 71.9% of the variation in lineage 2; b Distance plot for lineage 2 showing pairwise number of variant differences between samples; c Principal component 1 (PC1) by principal component 2 (PC2) for lineage 4, the first 10 principal components account for 88.9% of the variation in lineage 4. d Distance plot for lineage 2 showing pairwise number of variant differences between samples. (PPTX 5650 kb)
Scree plots for the principal component analyses, Scree plots showing the proportion of variation accounted for by the first ten principal components, calculated for the pairwise distances within a lineage 4 and b lineage 2. (PPTX 142 kb)
Drug-resistance phenotype frequency table, Drug-resistance phenotype frequency table by lineage. ‘Totals’ shows the number and percentage of each lineage with a known drug-resistance phenotype. (PPTX 45 kb)
Cross-resistance phenotype table, Cross-Resistance Table upper diagonal shows proportion of samples phenotyped for both vertical and horizontal phenotype, that test positive for vertical phenotype. Diagonal shows number of samples with each phenotype. Lower diagonal shows number of samples with phenotype for both horizontal and vertical phenotype. (PPTX 45 kb)
Variant Position Table, Table detailing variants at all positions with at least one non-synonymous variant found to be significantly associated with a phenotype in any of the variant-based analyses. (PPTX 52 kb)
Locus Comparison Table, Locus comparison table showing which analyses and in which lineage each loci was identified. An ‘x’ indicates a locus which was not identified by the method of analysis in question. Loci without a known association with the phenotype are highlighted in bold. (PPTX 44 kb)
Study frequency table, Study frequency table, showing numbers and percentage of strains from each study by lineage. (PPTX 40 kb)
Sublineage frequency table, Numbers and percentage by lineage assigned to each sublineage. (PPTX 36 kb)
Acknowledgments
The authors wish to thank the study participants.
Funding
YO and JPh are funded by a BBSRC PhD studentship (Grant no. BB/J014567/1). TC is funded by the Medical Research Council UK (Grant no. MR/M01360X/1, MR/N010469/1, MR/R025576/1, and MR/R020973/1) and BBSRC (Grant no. BB/R013063/1). The MRC eMedLab computing resource was used for bioinformatics and statistical analysis. Further funds were received from Fundação para a Ciência e a Tecnologia, Portugal, through the grants UID/Multi/04413/2013 (DM and MV). The funding bodies played no role in design of the study, collection, analysis and interpretation of data or in writing the manuscript.
Availability of data and materials
The analysis was performed on raw sequencing data available from the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena) under the following study accession numbers; PRJEB10385, ERP006619, ERP002611, ERP000192, SRP018402 and ERP008770, as utilized in Coll et al. 2017 [13].
Abbreviations
- AG
Aminoglycoside-resistance
- AMK
Amikacin-resistance
- CAP
Capreomycin-resistance
- CIP
Ciprofloxacin-resistance
- EMB
Ethambutol-resistance
- ETH
Ethionamide-resistance
- FQ
Fluoroquinolone-resistance
- GWAS
Genome-wide Association Study
- INH
Isoniazid-resistance
- KAN
Kanamycin-resistance
- MDR
Multidrug-resistant
- MOX
Moxifloxacin-resistance
- OFL
Ofloxacin-resistance
- PAN
Pan-susceptible; no known drug-resistance
- PAS
Para-Aminosalicylic Acid-resistance
- PCA
Principal Component Analysis
- PZA
Pyrazinamide-resistance
- RMP
Rifampicin-resistance
- STM
Streptomycin-resistance
- XDR
Extensively drug-resistant
Authors’ contributions
TC and MH conceived and directed the project. JPh generated the sequencing dataset. YO performed bioinformatic and statistical analyses under the supervision of TC and MH, and wrote the first draft of the manuscript. JPe, DM, AM, IP and MV contributed protocols and data. All authors commented and edited on various versions of the draft manuscript. All authors compiled and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yaa E. A. Oppong, Phone: +44 (0) 20 7636 8636, Email: yaa.oppong@lshtm.ac.uk
Jody Phelan, Email: jody.phelan@lshtm.ac.uk.
João Perdigão, Email: jrlperdigao@gmail.com.
Diana Machado, Email: diana@ihmt.unl.pt.
Anabela Miranda, Email: bsp1am@hotmail.com.
Isabel Portugal, Email: isabel.portugal@ff.ul.pt.
Miguel Viveiros, Email: mviveiros@ihmt.unl.pt.
Taane G. Clark, Email: taane.clark@lshtm.ac.uk
Martin L. Hibberd, Email: martin.hibberd@lshtm.ac.uk
References
- 1.World Health Organisation . Global Tuberculosis Report. 2015. p. 2015. [Google Scholar]
- 2.Mariam SH, Werngren J, Aronsson J, Hoffner S, Andersson DI. Dynamics of Antibiotic Resistant Mycobacterium tuberculosis during Long-Term Infection and Antibiotic Treatment. PLoS One. 2011;6:e21147. https://dx.plos.org/10.1371/journal.pone.0021147. [DOI] [PMC free article] [PubMed]
- 3.Fortune SM. The surprising diversity of mycobacterium tuberculosis: change you can believe in. J Infect Dis. 2012;206:1642–1644. doi: 10.1093/infdis/jis603. [DOI] [PubMed] [Google Scholar]
- 4.Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis. 2012;206:1724–1733. doi: 10.1093/infdis/jis601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark TG, Mallard K, Coll F, Preston M, Assefa S, Harris D, et al. Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment experienced patients by whole genome sequencing. PLoS One. 2013;8:1–12. doi: 10.1371/journal.pone.0083012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eldholm V, Monteserin J, Rieux A, Lopez B, Sobkowiak B, Ritacco V, et al. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat. Commun. 2015;6:7119. doi: 10.1038/ncomms8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015;7:51. doi: 10.1186/s13073-015-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller B, Borrell S, Rose G, Gagneux S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013;29:160–169. doi: 10.1016/j.tig.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Telenti A, Imboden P, Marchesi F, Matter L, Schopfer K, Bodmer T, et al. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet. 1993;341:647–651. doi: 10.1016/0140-6736(93)90417-F. [DOI] [PubMed] [Google Scholar]
- 10.Moradigaravand D, Grandjean L, Martinez E, Li H, Zheng J, Coronel J, et al. DfrA-thyA double deletion in para -aminosalicylic acid resistant Mycobacterium tuberculosis Beijing strains. Antimicrob. Agents Chemother. 2016:AAC.00253, 16 Available from: http://aac.asm.org/lookup/doi/10.1128/AAC.00253-16. [DOI] [PMC free article] [PubMed]
- 11.Galagan JE. Genomic insights into tuberculosis. Nat. Rev. genet. 2014;15:307–320. doi: 10.1038/nrg3664. [DOI] [PubMed] [Google Scholar]
- 12.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 2014;46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coll F, Phelan J, Hill-Cawthorne GA, Nair MB, Mallard K, Ali S, et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat Genet. 2018;50:307–316. doi: 10.1038/s41588-017-0029-0. [DOI] [PubMed] [Google Scholar]
- 14.Sveinbjornsson G, Gudbjartsson DF, Halldorsson B V, Kristinsson KG, Gottfredsson M, Barrett JC, et al. HLA class II sequence variants influence tuberculosis risk in populations of European ancestry. Nat. Genet. 2016;48:318–322. Available from: http://www.nature.com/doifinder/10.1038/ng.3498. [DOI] [PMC free article] [PubMed]
- 15.Cain AK, Lees JA. Using genomics to combat infectious diseases on a global scale. Genome Biol; 2015;16:250. Available from: http://genomebiology.com/2015/16/1/250 [DOI] [PMC free article] [PubMed]
- 16.Phelan J, Coll F, McNerney R, Ascher DB, Pires DE V., Furnham N, et al. Mycobacterium tuberculosis whole genome sequencing and protein structure modelling provides insights into anti-tuberculosis drug resistance. BMC Med; 2016;14:31. Available from: http://www.biomedcentral.com/1741-7015/14/31 [DOI] [PMC free article] [PubMed]
- 17.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Earle SG, Wu C, Charlesworth J, Stoesser N, Gordon NC, Walker TM, et al. Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat. Microbiol. 2016;1:16041. Available from: 10.1038/nmicrobiol.2016.41. [DOI] [PMC free article] [PubMed]
- 19.Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, Victor TC, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 2013;45:1183–9. Available from: http://www.nature.com/articles/ng.2747. [DOI] [PMC free article] [PubMed]
- 20.Desjardins CA, Cohen KA, Munsamy V, Abeel T, Maharaj K, Walker BJ, et al. Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat. Genet. 2016;48:1–9. doi: 10.1038/ng.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed MB, Pichler VK, Mcintosh F, Mattia A, Fallow A, Masala S, et al. Major mycobacterium tuberculosis lineages associate with patient country of origin. J Clin Microbiol. 2009;47:1119–1128. doi: 10.1128/JCM.02142-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2012;367:850–859. doi: 10.1098/rstb.2011.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Click ES, Moonan PK, Winston CA, Cowan LS, Oeltmann JE. Relationship between mycobacterium tuberculosis phylogenetic lineage and clinical site of tuberculosis. Clin Infect Dis. 2012;54:211–219. doi: 10.1093/cid/cir788. [DOI] [PubMed] [Google Scholar]
- 24.Krishnan N, Malaga W, Constant P, Caws M, Thi Hoang Chau T, Salmons J, et al. Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PLoS One. 2011;6. [DOI] [PMC free article] [PubMed]
- 25.Portevin D, Gagneux S, Comas I, Young D. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. 2011;7. [DOI] [PMC free article] [PubMed]
- 26.Mathema B, Kurepina N, Yang G, Shashkina E, Manca C, Mehaffy C, et al. Epidemiologic consequences of microvariation in Mycobacterium tuberculosis. J Infect Dis. 2012;205:964–974. doi: 10.1093/infdis/jir876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 2016; Available from: http://www.nature.com/doifinder/10.1038/ng.3704. [DOI] [PMC free article] [PubMed]
- 28.Coll F, McNerney R, Guerra-Assunção JA, Glynn JR, Perdigão J, Viveiros M, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014;5:4812. Available from: http://www.nature.com/articles/ncomms5812. [DOI] [PMC free article] [PubMed]
- 29.Benavente ED, Coll F, Furnham N, McNerney R, Glynn JR, Campino S, et al. PhyTB: Phylogenetic tree visualisation and sample positioning for M. tuberculosis. BMC Bioinformatics. 2015;16:155. Available from: http://www.biomedcentral.com/1471-2105/16/155. [DOI] [PMC free article] [PubMed]
- 30.Perdigão J, Macedo R, Machado D, Silva C, Jordão L, Couto I, et al. GidB mutation as a phylogenetic marker for Q1 cluster Mycobacterium tuberculosis isolates and intermediate-level streptomycin resistance determinant in Lisbon, Portugal. Clin. Microbiol. Infect. 2014;20. [DOI] [PubMed]
- 31.Machado D, Perdigão J, Ramos J, Couto I, Portugal I, Ritter C, et al. High-level resistance to isoniazid and ethionamide in multidrug-resistant Mycobacterium tuberculosis of the Lisboa family is associated with inhA double mutations. J Antimicrob Chemother. 2013;68:1728–1732. doi: 10.1093/jac/dkt090. [DOI] [PubMed] [Google Scholar]
- 32.Guerra-Assunção J, Crampin A, Houben R, Mzembe T, Mallard K, Coll F, et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife. 2015;4:1–17. doi: 10.7554/eLife.05166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, et al. High Functional Diversity in Mycobacterium tuberculosis Driven by Genetic Drift and Human Demography. Blaser MJ, editor. PLoS Biol. 2008;6:e311. Available from: https://dx.plos.org/10.1371/journal.pbio.0060311. [DOI] [PMC free article] [PubMed]
- 34.Dong Y, Qiu X, Shaw N, Xu Y, Sun Y, Li X, et al. Molecular basis for the inhibition of β-hydroxyacyl-ACP dehydratase HadAB complex from mycobacterium tuberculosis by flavonoid inhibitors. Protein Cell Higher Education Press. 2015;6:504–517. doi: 10.1007/s13238-015-0181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gannoun-Zaki L, Alibaud L, Kremer L. Point mutations within the fatty acid synthase type II dehydratase components HadA or HadC contribute to isoxyl resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57:629–632. doi: 10.1128/AAC.01972-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nebenzahl-Guimaraes H, Van Laarhoven A, Farhat MR, Koeken VACM, Mandemakers JJ, Zomer A, et al. Transmissible mycobacterium tuberculosis strains share genetic markers and immune phenotypes. Am J Respir Crit Care Med. 2017;195:1519–1527. doi: 10.1164/rccm.201605-1042OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Hu Y, Cumming BM, Lu P, Feng L, Deng J, et al. Mycobacterial WhiB6 Differentially Regulates ESX-1 and the Dos Regulon to Modulate Granuloma Formation and Virulence in Zebrafish. Cell Rep. The Author(s); 2016;16:2512–2524. Available from: 10.1016/j.celrep.2016.07.080 [DOI] [PubMed]
- 38.Solans L, Aguiló N, Samper S, Pawlik A, Frigui W, Martín C, et al. A specific polymorphism in Mycobacterium tuberculosis H37Rv causes differential ESAT-6 expression and identifies whiB6 as a novel ESX-1 component. Infect Immun. 2014;82:3446–3456. doi: 10.1128/IAI.01824-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen T, He L, Deng W, Xie J. The Mycobacterium DosR regulon structure and diversity revealed by comparative genomic analysis. J Cell Biochem. 2013;6:1–6. doi: 10.1002/jcb.24302. [DOI] [PubMed] [Google Scholar]
- 40.Domenech P, Zou J, Averback A, Syed N, Curtis D, Donato S, et al. Unique regulation of the DosR regulon in the Beijing lineage of Mycobacterium tuberculosis. J Bacteriol. 2017;199:1–19. doi: 10.1128/JB.00696-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi T, Xie J. Molybdenum enzymes and molybdenum cofactor in mycobacteria. J Cell Biochem. 2011;112:2721–2728. doi: 10.1002/jcb.23233. [DOI] [PubMed] [Google Scholar]
- 42.Levillain F, Poquet Y, Mallet L, Mazères S, Marceau M, Brosch R, et al. Horizontal acquisition of a hypoxiaresponsive molybdenum cofactor biosynthesis pathway contributed to Mycobacterium tuberculosis pathoadaptation. PLOS Pathog. 2017;13:e1006752. Available from: https://dx.plos.org/10.1371/journal.ppat.1006752. [DOI] [PMC free article] [PubMed]
- 43.Shah NS, Auld SC, Brust JCM, Mathema B, Ismail N, Moodley P, et al. Transmission of extensively drug-resistant tuberculosis in South Africa. N Engl J Med [Internet] 2017;376:243–253. doi: 10.1056/NEJMoa1604544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perdigão J, Silva H, Machado D, Macedo R, Maltez F, Silva C, et al. Unraveling Mycobacterium tuberculosis genomic diversity and evolution in Lisbon, Portugal, a highly drug resistant setting. BMC Genomics. 2014;15:991. doi: 10.1186/1471-2164-15-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics [Internet]. 2009;25:2078–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19505943. [DOI] [PMC free article] [PubMed]
- 47.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozlov AM, Aberer AJ, Stamatakis A. ExaML version 3: a tool for phylogenomic analyses on supercomputers. Bioinformatics. 2015;31:2577–2579. doi: 10.1093/bioinformatics/btv184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Letunic I, Bork P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 50.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. Nature Publishing Group; 2012;44:821–824. Available from: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed]
- 51.Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018;3:731. Available from: http://joss.theoj.org/papers/10.21105/joss.00731.
- 52.Core R. Team. R: a language and environment for statistical computing. [internet]. Vienna, Austria: R Foundation for Statistical. Computing. 2015; Available from: https://www.r-project.org/.
- 53.Torres JN, Paul LV, Rodwell TC, Victor TC, Amallraja AM, Elghraoui A, et al. Novel katG mutations causing isoniazid resistance in clinical M. tuberculosis isolates. Emerg. Microbes Infect. 2015;4:e42. doi: 10.1038/emi.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Variant Summary Tables, Summary tables of variants called in comparison to the H37rv reference, with monomorphic variants removed for each dataset. a Total numbers of variants by lineage; b Number of variants per sample; c Non-reference variant frequency summary; variants called in comparison to the H37rv reference. (PPTX 39 kb)
Non-reference variant frequency histogram, A histogram showing log10(frequency + 1) of non-reference alleles compared to the H37rv reference for a lineage 2 and b lineage4. (PPTX 69 kb)
Population diversity within investigated strains, a Principal component 1 (PC1) by principal component 2 (PC2) for lineage 2, The first 10 principal components account for 71.9% of the variation in lineage 2; b Distance plot for lineage 2 showing pairwise number of variant differences between samples; c Principal component 1 (PC1) by principal component 2 (PC2) for lineage 4, the first 10 principal components account for 88.9% of the variation in lineage 4. d Distance plot for lineage 2 showing pairwise number of variant differences between samples. (PPTX 5650 kb)
Scree plots for the principal component analyses, Scree plots showing the proportion of variation accounted for by the first ten principal components, calculated for the pairwise distances within a lineage 4 and b lineage 2. (PPTX 142 kb)
Drug-resistance phenotype frequency table, Drug-resistance phenotype frequency table by lineage. ‘Totals’ shows the number and percentage of each lineage with a known drug-resistance phenotype. (PPTX 45 kb)
Cross-resistance phenotype table, Cross-Resistance Table upper diagonal shows proportion of samples phenotyped for both vertical and horizontal phenotype, that test positive for vertical phenotype. Diagonal shows number of samples with each phenotype. Lower diagonal shows number of samples with phenotype for both horizontal and vertical phenotype. (PPTX 45 kb)
Variant Position Table, Table detailing variants at all positions with at least one non-synonymous variant found to be significantly associated with a phenotype in any of the variant-based analyses. (PPTX 52 kb)
Locus Comparison Table, Locus comparison table showing which analyses and in which lineage each loci was identified. An ‘x’ indicates a locus which was not identified by the method of analysis in question. Loci without a known association with the phenotype are highlighted in bold. (PPTX 44 kb)
Study frequency table, Study frequency table, showing numbers and percentage of strains from each study by lineage. (PPTX 40 kb)
Sublineage frequency table, Numbers and percentage by lineage assigned to each sublineage. (PPTX 36 kb)
Data Availability Statement
The analysis was performed on raw sequencing data available from the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena) under the following study accession numbers; PRJEB10385, ERP006619, ERP002611, ERP000192, SRP018402 and ERP008770, as utilized in Coll et al. 2017 [13].