

Abstract
Purpose:
Tumor-infiltrating immune cells have prognostic significance and are attractive therapeutic targets. Yet, the clinical significance of their spatial organization and phenotype in diffuse large B-cell lymphoma (DLBCL) is unclear.
Experimental Design:
We characterized T cells, macrophages, and their spatial interactions by multiplex IHC (mIHC) in 178 patients with DLBCL and correlated the data with patient demographics and survival. We validated the findings on gene expression data from two external DLBCL cohorts comprising 633 patients.
Results:
Macrophage and T-cell contents divided the samples into T cell–inflamed (60%) and noninflamed (40%) subgroups. The T cell–inflamed lymphoma microenvironment (LME) was also rich in other immune cells, defining immune hot phenotype, which did not as such correlate with outcome. However, when we divided the patients according to T-cell and macrophage contents, LME characterized by high T-cell/low macrophage content or a corresponding gene signature was associated with superior survival [5-year overall survival (OS): 92.3% vs. 74.4%, P = 0.036; 5-year progression-free survival (PFS): 92.6% vs. 69.8%, P = 0.012]. High proportion of PD-L1- and TIM3-expressing CD163− macrophages in the T cell–inflamed LME defined a group of patients with poor outcome [OS: HR = 3.22, 95% confidence interval (CI), 1.63–6.37, Padj = 0.011; PFS: HR = 2.76, 95% CI, 1.44–5.28, Padj = 0.016]. Furthermore, PD-L1 and PD-1 were enriched on macrophages interacting with T cells.
Conclusions:
Our data demonstrate that the interplay between macrophages and T cells in the DLBCL LME is immune checkpoint dependent and clinically meaningful.
Translational Relevance.
Unlike in inflamed lymphomas, such as classical Hodgkin lymphoma (cHL), responses to immune checkpoint blockade therapy (CBT) have been low in diffuse large B-cell lymphoma (DLBCL). Here we characterize DLBCL microenvironment (LME) on a cellular level, extending beyond the gene expression level, which has been the focus of previous studies. We show that tumor infiltrating T cells and macrophages divide DLBCLs into inflamed and noninflamed subgroups and that their proportions, immunophenotypes, and spatial interactions in the LME have prognostic impact on the survival in patients with DLBCL. Together, our results demonstrate that the interplay between different immune cells in the LME is clinically more meaningful than the proportions of single-cell subtypes alone. In particular, the macrophage content seems to impair the antitumoral activity of T cells, possibly by immune checkpoint–related mechanisms. We speculate that inflamed DLBCLs with a high proportion of macrophages are potential targets of CBT.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoid malignancy in adults. Approximately two thirds of the patients are cured in response to standard rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP)-based immunochemotherapy (1). Based on the cell of origin, DLBCL is divided into germinal center B-cell like (GCB) and activated B-cell like (ABC) subgroups (2), which are addicted to different oncogenic pathways and differ in their clinical course (3, 4). In addition, novel genomic entities, which can better delineate GCB and ABC subgroups have recently been identified (5–7). To date, however, subtype-targeted therapies have not been superior to R-CHOP regimen (8, 9).
Beyond tumor cell–derived factors, clinical course of DLBCL is impacted by the tumor microenvironment (10–15). B-cell lymphoma microenvironment (LME) is heterogeneous, consisting of blood vessels, extracellular matrix, stromal cells, and immune cells, including T cells, macrophages, and natural killer (NK) cells (16). LME can constitute an immune response against cancer cells (16). However, it can also support lymphoma cells by inhibiting T-cell infiltration into lymphoma tissues or by preventing T-cell priming (17). It can also suppress T cells already present in the lymphoma by inducing exhaustion (18). In summary, LME can create an immune compromised milieu allowing malignant cells to proliferate without a risk of being detected by the immune system.
Based on T-cell infiltration, lymphomas can be divided into T cell–inflamed and non-T cell–inflamed phenotypes (19). In addition to harboring rich T-cell infiltrates, inflamed lymphomas are characterized by overactivation of NF-κB signaling (20, 21), and promotion of immune evasion mechanisms, resulting in downregulation of human leukocyte antigen (HLA) I and II, which are needed for effective T-cell detection (16, 22–25). Recruitment of immune-suppressive macrophages and upregulation of immune checkpoint molecules, such as programmed death-1 (PD-1) and PD-ligand-1 (PD-L1) are other common immune evasion techniques of inflamed lymphomas (26–28). Immune checkpoint molecules can modify active immune cells to become inactive, and suppress the immune response against the tumor (18). PD-1–targeting checkpoint blockade therapies (CBT) have shown promising response rates in inflamed lymphomas, such as classical Hodgkin lymphoma (cHL), and primary mediastinal large B-cell lymphoma (PMBL; refs. 29, 30).
Conversely, noninflamed lymphomas are mostly devoid of immune cells. This may be because of the alterations preventing immune cell recruitment, or because of physical exclusion of immune cells from the LME (31). DLBCL has traditionally been classified as a non-inflamed lymphoma (19). It appears to be more heterogeneous in its LME constitution than previously anticipated, and in some DLBCLs an anti-lymphoma immune response can be triggered (32, 33). Recently, subtyping of DLBCL based on the composition of the LME has been proposed (14). Several genetic alterations have also been found to correlate with inflamed and noninflamed LME (23, 33, 34). Unlike in cHL, responses to CBT are scarce in relapsed/refractory (r/r) DLBCL (35), and the identification of patients benefitting from CBT remains a challenge.
Previously, we studied the clinical significance of immune checkpoint–expressing T cells in the DLBCL microenvironment, and observed that a high proportion of TIM3+ T cells at diagnosis predicted poor outcome (13). Here, we sought to investigate the network between T cells, macrophages, and NK cells in the DLBCL microenvironment. We also addressed the impact of immune checkpoint molecules on the relations between these cells.
Materials and Methods
Patients and samples
A flowchart showing the patient cohorts used in this study is presented in Supplementary Fig. S1. Multiplex IHC (mIHC) cohort consisted of 178 patients diagnosed with primary DLBCL and treated with immunochemotherapy. Tissue microarrays (TMA) were constructed from formalin-fixed paraffin-embedded (FFPE) lymphoma samples. RNA-sequencing data were available from 77 patients, and digital multiplexed gene expression profiling data (NanoString nCounter Human PanCancer Immunoprofiling Panel) from 38 of the patients (13). Patients and corresponding clinical data were retrospectively extracted from electronic and/or paper-based medical records. For validation, we used gene expression data from Reddy and colleagues (EGAS00001002606; n = 624) and Chapuy and colleagues (GSE98588; n = 137) cohorts (5, 7). In the Reddy and colleagues cohort, 128 patients overlapping with the mIHC cohort were excluded from the original cohort, resulting in 496 patients. Patient data were handled according to Good Scientific Practice (GSP) Guidelines. The study was approved by the Ethics Committee in Helsinki University Hospital and by the Finnish National Authority for Medicolegal Affairs, which waived the requirement to obtain informed consent.
IHC
We performed mIHC on TMAs to characterize macrophage, B-cell, T-cell, and NK-cell immunophenotypes (panel 1: CD68, CD163, CD20, PD-L1, TIM3; Panel 2: CD68, CD163, CD3, CD4, PD-1, PD-L1; Panel 3: CD56, CD3, CD45, TIM3, Granzyme B) using the protocol described earlier, with some modifications (Supplementary Table S1; ref. 36). Two targets were detected using Alexa Fluor 488 and Alexa Fluor 555 channels amplified using tyramide signal amplification (TSA; PerkinElmer). A pair of primary antibodies raised in different species was used to detect another two targets using Alexa Fluor 647 and Alexa Fluor 750 fluorochrome-conjugated secondary antibodies without amplification. We counterstained nuclei using DAPI and mounted and applied coverslips on the slides. After first-round staining and whole-slide imaging of the TMAs, we removed the coverslips by soaking the slides in wash buffer at 4°C. We then soaked the slides in TBS buffer containing 25 mmol/L NaOH and 4.5% H2O2 to bleach the previous Alexa Fluor staining. Finally, we heated the slides in 1 mmol/L Tris/10 mmol/L EDTA pH 9 solution for 20 minutes at 99°C to denature the antibodies from the first-round staining. We then preformed a second-round staining to detect one or two additional targets using Alexa Fluor 647 and Alexa Fluor 750 secondary antibodies.
Imaging
We acquired digital, fluorescence images of mIHC slides using Zeiss Axio Scan.Z1 with Zeiss 20X (0.8NA, M27) Plan-Apochromat objective, Hamamatsu ORCA-Flash 4.0 V2 Digital CMOS camera (16-bit; 0.325 μm/pixel resolution), and Zeiss Colibri.7 LED Light Source. We used the following filter specifications: DAPI cube (Zeiss Filter Set 02), FITC cube (Zeiss Filter Set 38 HE), Cy3 cube (Chroma Technology Corp 49004 ET CY3/R), Cy5 cube (Chrome Technology Corp 49006 ET CY5), Cy7 cube (Chroma Technology Corp 49007 ET CY7). After image acquisition, we converted images to 8-bit JPEG2000 format (100% quality).
Image analysis
We used the Ilastik v.1.3.3 software to filter out areas with staining artefacts due to autofluorescence (37). We performed automated quantification using CellProfiler v.3.1.8 software, as described previously (13, 38). We determined cell classes using pixel colocalization analysis. Each channel intensity was thresholded using Adaptive Otsu. We determined double or triple channel positive pixels with “MaskImage”, determined thresholded channel pixel areas with “MeasureImageAreaOccupied” and counted areal proportions by dividing the area with pixel area occupied by all the channels combined (ImageMath Add command). We exported cell class areas as CSV files with “ExportToSpreadsheet”. By visual inspection, we determined the quality of TMA cores and excluded low quality cores (e.g., ruptured or folded tissue) from further analyses. Furthermore, we excluded some samples from specific clustering and survival analyses due to staining artefacts. We also utilized data from proportions of CD8+, FoxP3+, and LAG3+ cells from previously performed mIHC analyses (13). Finally, we utilized data on the expression of MYC, BCL2, HLA-ABC, and HLA-DR from previously performed IHC analyses (13, 39). Cases with overexpression of both MYC and BCL2 were classified as double protein expressors (DPE).
In silico immunophenotyping
We performed CIBERSORTx deconvolution analyses utilizing the web portal (http://cibersortx.stanford.edu) on gene expression data from the Reddy and colleagues cohort to infer the proportions of tumor-infiltrating immune cell subtypes and their gene expressions. We ran the algorithm using the 547-gene Leukocyte gene signature matrix (LM22) with 100 permutations as described by Newman and colleagues (40, 41).
Differentially expressed genes and pathway analyses
For the differential gene expression analyses, we used the R/Bioconductor package limma (42). The cut-off for differentially expressed (DE) genes in the T-cell high/macrophage-low group compared to the rest was FDR-adjusted P value < 0.001 and absolute log2 fold change (log2FC) > 0.9. We used single-sample Gene Set Enrichment Analysis (ssGSEA) to calculate scores for the specific pathways in the KEGG, Reactome, and Biocarta databases for every individual sample (43). The differential pathway scores between the T cell–high and T cell–low groups, as well as between the T cell high/macrophage low and T cell high/macrophage high or T cell low groups were compared by Mann-Whitney U test with FDR correction.
Cell interaction analyses
We performed cell-cell interaction analyses using the method developed by Brück and colleagues (44). First, we performed cell segmentation of the mIHC-stained tissue samples using the CellProfiler software, and identified the phenotype of each individual cell. For segmentation we thresholded each channel intensity using Adaptive Otsu. Clumped objects were separated based on intensity. The distance between two cells was calculated as the Euclidean distance between the center points of the cells. Cells were defined to interact when they were closer than 100 pixels (22 μm) from each other (45). The number of interacting cells in each sample was normalized to the total number of cells by calculating an interaction index Iab using the formula:
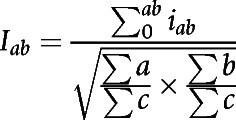 |
where iab is the interaction between any two cells a and b, ∑a is the sum of a cells, ∑b is the sum of b cells, and ∑c is the sum of all cells in the sample. The fold change (FC) between the expression of immune checkpoint molecules in cells a when they interacted with cells b compared to when they did not, was calculated as the median number of immune checkpoint positive cells a interacting with cells b divided by the median number of noninteracting immune checkpoint positive cells a in all samples. Finally, the difference between the proportion of cells a expressing immune checkpoint molecules when they interacted with cells b between any two groups 1 and 2 was calculated as the median of the proportion of cells a expressing immune checkpoint molecules when they interacted with cells b. The FC was then calculated by dividing the median in group 1 with the median in group 2.
Statistical analyses
We performed all statistical analyses with R v.4.0.1. We performed unsupervised hierarchical clustering for z-score normalized data with the pheatmap package using Euclidean distance with Ward's linkage. We estimated the prognostic impact of variables using univariate and multivariate Cox regression models. We estimated the difference in survival between patient groups using the Kaplan–Meier method with log-rank test. The median follow-up time was calculated by reverse Kaplan–Meier estimator. OS was defined as the time from diagnosis to death from any cause and PFS as the time from diagnosis to progression or death from any cause. To compare two or more groups, we used Mann–Whitney U and Kruskal–Wallis H tests, respectively. We corrected P-values for the errors due to multiple testing using the FDR method.
Data availability
The data generated in this study are available upon request from the corresponding author.
Results
DLBCL microenvironment is heterogeneous
Our mIHC cohort consisted of 178 patients with DLBCL treated with R-CHOP or R-CHOP-like immunochemotherapy (Table 1). The median follow-up time was 63 months [interquartile range (IQR): 48 months–69 months], during which 33 patients relapsed and 37 died, translating to 74% progression-free survival (PFS) and 78% overall survival (OS) rates at 5 year, respectively. Of the established risk factors, International Prognostic Index (IPI) score, molecular subtype, and MYC and BCL2 double-protein expression (DPE) associated with unfavorable survival (Supplementary Table S2).
Table 1.
Patient demographics.
| Characteristics | mIHC cohort, n (%) | Reddy et al. cohort, n (%) |
|---|---|---|
| No. of patients | 178 (100) | 496 (100) |
| Age | ||
| Median (range) | 60 (16–84) | |
| <60 | 88 (49) | 205 (43) |
| ≥60 | 90 (51) | 268 (57) |
| ND | 23 | |
| Gender | ||
| Male | 103 (58) | 270 (54) |
| Female | 75 (42) | 226 (46) |
| Molecular subtype | ||
| GCB | 62 (45) | 217 (44) |
| ABC | 55 (40) | 203 (41) |
| Unclassified | 21 (15) | 76 (15) |
| ND | 40 | |
| WHO PS | ||
| 0–1 | 125 (71) | 349 (76) |
| ≥2 | 50 (29) | 112 (24) |
| ND | 3 | 35 |
| Stage | ||
| I–II | 77 (44) | 179 (37) |
| III–IV | 100 (56) | 307 (63) |
| ND | 1 | 10 |
| IPI | ||
| 0–2 | 91 (52) | 212 (55) |
| 3–5 | 84 (48) | 176 (45) |
| ND | 3 | 108 |
| Elevated LDH | ||
| Yes | 113 (65) | 235 (53) |
| No | 62 (35) | 206 (47) |
| ND | 3 | 55 |
| EN | ||
| 0–1 | 122 (73) | 341 (73) |
| ≥2 | 46 (27) | 123 (27) |
| ND | 10 | 32 |
| B-symptoms | ||
| Yes | 65 (38) | |
| No | 108 (62) | |
| ND | 5 | |
| Double expressor | ||
| Yes | 49 (34) | |
| No | 97 (66) | |
| ND | 32 | |
| Double hit | ||
| Yes | 8 (5) | |
| No | 141 (95) | |
| ND | 29 | |
| Treatment | ||
| R-CHOP | 116 (65) | |
| R-CHOEPa | 58 (33) | |
| Otherb | 4 (2) | |
| 5-year PFS | 74% | |
| 5-year OS | 78% | 63% |
Abbreviations: ABC, activated B-cell; GCB, germinal center B; IPI, international prognostic index; LDH, lactate dehydrogenase; EN, extranodal site; R-CHOP, rituximab, cyclophosphamide, doxorubicine, vincristine, prednisone; R-CHOEP, R-CHOP + etoposide.
aPatients <65 years with high-risk features were treated with R-CHOEP-14 and systemic CNS prophylaxis consisting of high-dose methotrexate and high-dose cytarabine.
bOther, one R-CHOP/R-CHEP, one R-CHOP/R-CNOP, one R-CHOP/R-mini-CHOP, one R-ICE (excluded from survival analyses).
First, we characterized the composition of LME by analyzing macrophage, B-cell, T-cell, and NK-cell proportions and their immunophenotypes (Fig. 1A; Supplementary Fig. S2; Supplementary Table S3). We found high variability between the samples in terms of LME cell composition. The median proportion of T cells was 15.1% (0.4%–61.5%), macrophages 9.9% (0.6%–23.8%), and B cells 60.4% (0.1%–92.2%). The proportion of NK cells was generally low with a median of 0.004% (0%–0.3%), most samples having no or very few NK cells in their LME (Fig. 1B). The most abundant immune checkpoint molecule was PD-1 (Fig. 1C), the expression of which was prominent especially on T cells, while PD-L1 expression was more abundant on macrophages (Fig. 1C–F). The proportions of cells expressing the studied markers correlated well with the corresponding gene expression (Supplementary Fig. S3).
Figure 1.

The heterogeneity of the DLBCL microenvironment revealed by mIHC. A, Representative images from mIHC analyses performed on TMAs. Macrophages and B cells: CD20, blue; CD68, green; CD163, magenta; TIM3, red; PD-L1, yellow; DAPI, gray. Macrophages and T cells: CD3, blue; CD4, cyan; CD68, green; CD163, magenta; PD-1, red; PD-L1, yellow; DAPI, gray. NK cells: CD45, magenta; CD3, blue; CD56, yellow; TIM3, red; Granzyme B, green; DAPI, gray. Scale bar, 50 μm. B and C, Boxplots depicting the proportions of different immune cell subtypes (B) and immune checkpoint–expressing cells (C) out of all cells in the DLBCL LME. D–F, Boxplots depicting the proportions of T cells (D), macrophages (E), and NK cells (F) expressing immune checkpoint molecules in the DLBCL LME. G, Unsupervised hierarchical clustering of all quantified immune cells and their immunophenotypes. Heat map represents z-score normalized data; red color denotes expression above mean, blue color expression below mean, and white color mean. Gray color denotes missing data.
To explore the heterogeneity of the LME, we performed unsupervised hierarchical clustering for the mIHC cohort with all the studied cell phenotypes (Fig. 1G). DLBCL samples clustered into immune cell high (inflamed) and low (noninflamed) LME subgroups. Within the inflamed LME group, samples further formed smaller clusters, which were characterized by high proportions of cells expressing one or more immune checkpoint molecule, high proportion of T cells, or high proportion of macrophages. Finally, few samples were characterized by high proportion of NK cells. The samples with high proportion of B cells were generally associated with fewer T and NK cells, as well as macrophages.
Clinical impact of immune cells in the LME
Next, we performed unsupervised hierarchical clustering separately for macrophages, NK cells, and T cells. According to the proportion of macrophages, the samples formed three separate clusters: macrophage low, CD163+ (M2-like) macrophage high, and CD163− (M1-like) macrophage high clusters (Supplementary Fig. S4A). According to the NK-cell proportion, samples clustered into NK cell–low and -high groups (Supplementary Fig. S4B). Finally, approximately half of the samples had high T-cell content (T cell-inflamed; Fig. 2A), as previously reported (13). The T cell–inflamed group could further be divided into T-cell intermediate and T cell–high groups. None of the immune cell clusters correlated with outcome (data not shown).
Figure 2.

The clinical impact of T cells and macrophages in the DLBCL microenvironment. A, Unsupervised hierarchical clustering of T-cell subtypes quantified by mIHC. B–D, Boxplots depicting the proportions of macrophages (B), NK cells (C), and regulatory T cells (D) in the groups with high and low proportion of T cells in the mIHC cohort. E, Unsupervised hierarchical clustering of macrophage subtypes in the T cell–high group quantified by mIHC. F and G, Kaplan–Meier (log-rank test) survival plots depict OS in the T cell–high/macrophage-low (T cell–high/M low) and T cell–high/macrophage-high (T cell–high/M high) or T cell–low groups (F), as well as in the T cell–high/macrophage-low (T cell–high/M low), T cell–high/macrophage-high (T cell–high/M high), and the T cell–low groups (G) in the mIHC cohort. H, Unsupervised hierarchical clustering of T-cell subtypes in the Reddy and colleagues' cohort inferred by CIBERSORT. I, Unsupervised hierarchical clustering of macrophage subtypes in the T cell–high group in the Reddy and colleagues' cohort inferred by CIBERSORT. J and K, Kaplan–Meier (log-rank test) survival plots depict OS in the T-cell high/macrophage-low (T cell–high/M low), T cell–high/macrophage-high (T cell–high/M high), and T cell–low groups (J), as well as in the T cell–high/macrophage-low (T cell–high/M low) and T cell–high/macrophage high (T cell–high/M high) or T cell–low groups (K) in the Reddy and colleagues' cohort. L, Forest plot visualizing the impact of the T cell–high/macrophage-low group on OS in a Cox regression multivariate analysis with IPI and molecular subtype in the Reddy and colleagues' cohort. In A, E, G, and H, heat maps represent z-score normalized data; red color denotes expression above mean, blue color expression below mean, and white color mean. Gray color denotes missing data.
We found that DLBCLs with a T cell–inflamed microenvironment generally had high proportions of other immune cells, including macrophages, NK cells, and regulatory T cells (Tregs), as well as cells expressing immune checkpoint molecules PD-1, TIM3, PD-L1, and LAG3 (Fig. 2B–D; Supplementary Fig. S4C–S4O). Interestingly, T cell-inflamed DLBCLs differed in the proportion of macrophages, resulting in identification of a T cell–high/macrophage-low group (Fig. 2E). Patients with T cell–high/macrophage-low LME had a less aggressive disease defined by lower IPI scores and the GCB phenotype (Supplementary Table S4). This correlated with better survival (5-year OS: 92.3% vs. 74.4%, P = 0.038; 5-year PFS: 92.6% vs. 69.8%, P = 0.012; Fig. 2F and G; Supplementary Fig. S4P). T cell-noninflamed group could also be divided according to the proportion of macrophages (Supplementary Fig. S5A). However, no correlation with survival was found between T cell–low/macrophage-low and T cell–low/macrophage-high groups (Supplementary Fig. S5B–S5C). When double-hit lymphomas were excluded from the survival analyses, the prognostic impact of macrophages on survival in the T cell–inflamed and noninflamed groups remained unchanged (Supplementary Fig. S6). Finally, we studied the proportion between macrophages and T cells in the LME. In accordance with the positive prognostic impact of the T cell–high/macrophage-low group, the patients with low macrophage/T-cell proportion had a better OS and PFS than the patients with high macrophage/T-cell proportion (5-year OS: 81.7% vs. 62.0%, P = 0.0015; 5-year PFS: 76.9% vs. 62.0%, P = 0.013; Supplementary Fig. S7).
Validation of the mIHC data with CIBERSORT deconvolution analyses
Next, we utilized gene expression data from a validation cohort of 624 patients (Table 1), and estimated the immune cell proportions by CIBERSORT deconvolution analysis. When using data from the 77 samples overlapping with the mIHC cohort, distinct immune cell subtypes (Supplementary Table S5) correlated well with the mIHC data, with the exception of Tregs and NK cells, which were driven by outliers (Supplementary Fig. S8). In concordance with the mIHC cohort, unsupervised hierarchical clustering of the 496 nonoverlapping samples separated the patients into subgroups with high and low T cell (Fig. 2H), high and low macrophage, and high and low NK-cell proportions (Supplementary Fig. S9A and S9B). While no correlation between the proportions of macrophages nor NK cells and survival was seen, high proportion of T cells in the LME translated to better survival (5-year OS: 65.4% vs. 58.7%, P = 0.040, Supplementary Fig. S9C). As in our mIHC cohort, DLBCLs with high proportion of T cells displayed higher proportions of other immune cells, such as macrophages, NK cells, and Tregs, and higher gene expression of immune checkpoint molecules, such as PDCD1, HAVCR2, CD274, and LAG3, suggesting an immune-inflamed phenotype (Supplementary Fig. S9D–S9Q). Finally, when we clustered the T cell–high samples according to the proportion of macrophages, we identified a group with low number of macrophages (Fig. 2I), which associated with less aggressive clinical course (Supplementary Table S4), and better survival when compared with the patients with T cell–high and macrophage-high, or T cell–low LME (5-year OS: 69.7% vs. 58.3%, P = 0.004), thus, validating our initial findings (Fig. 2J and K). The impact of T cell–high/macrophage low LME on survival was independent of the IPI and molecular subtype (Fig. 2L).
Differential pathway enrichment between the distinct LME phenotypes
To study the differences in the cellular pathways between the distinct T-cell and macrophage groups, we performed ssGSEA. First, we used the RNA-sequencing data from 77 patients from the mIHC cohort to determine specific pathway scores for each individual sample, and then calculated differences in the pathway scores between T cell–high and low LME subgroups. As expected, the highest ranking pathways in the T cell high LME group were pathways related to T cells and T-cell signaling (Fig. 3A; Supplementary Table S6). On the contrary, the highest ranking pathways in the T cell–low LME group were the pathways related to cell division and DNA repair, perhaps representing greater cell division activity of the malignant B cells and a disease that is more independent from the surrounding LME. These findings were replicated in the Reddy and colleagues (5) validation cohort (Supplementary Fig. S10A; Supplementary Table S7).
Figure 3.

Differentially enriched pathways and differentially expressed genes between distinct LME phenotypes. A, A volcano plot depicting differentially enriched pathways between the T cell–high (inflamed) and T cell–low (noninflamed) groups in the mIHC cohort. B and C, Volcano plots depicting differentially enriched pathways (B) and differentially expressed genes (C) between the T cell–high/macrophage-low (T cell–high/M low) and T cell–high/macrophage-high or T cell–low groups in the Reddy and colleagues' cohort. D, Unsupervised hierarchical clustering of samples in the Reddy and colleagues' cohort based on the expression of the 18-gene signature most differentially expressed between the T-cell high/macrophage-low and T cell–high/macrophage-high or T cell–low groups. E, Kaplan–Meier (log-rank test) survival plot depicts OS in the groups with high and low expression of the T cell–high/macrophage-low gene signature in the Reddy and colleagues' cohort. F, Unsupervised hierarchical clustering of samples in the Chapuy and colleagues' cohort based on the expression of the 18-gene signature most differently expressed between the T cell–high/macrophage-low and T cell–high/macrophage-high or T cell–low groups. TRBC2 was not expressed in this cohort. G, Kaplan–Meier (log-rank test) survival plot depicts OS in the groups with high and low expression of the T cell–high/macrophage-low gene signature in the Chapuy and colleagues' cohort. In D and F, heat maps represent z-score normalized data; red color denotes expression above mean, blue color expression below mean, and white color mean. Gray color denotes missing data.
We also studied how the T cell–high/macrophage low LME subgroup differed from the rest of the samples and the T cell–high/macrophage-high subgroup in the Reddy and colleagues validation cohort and found that the highest ranking pathways were related to T-cell signaling, but also to B-cell receptor signaling (Fig. 3B; Supplementary Fig. S10B; Supplementary Tables S8 and S9). In contrast, the lowest ranking pathways were related to cell division and DNA repair.
Gene expression signature representing T cell–high/macrophage-low LME
To define a gene expression signature corresponding to the T cell–high/macrophage-low LME, we analyzed differentially expressed genes between the T cell–high/macrophage-low LME group and the others in the Reddy and colleagues cohort (5). The most upregulated genes in the T cell–high/macrophage-low LME group were genes related to T cells, such as CD3D, CD7, and CD28, but also other genes, such as BCL11B, CCL21, and SH2D1 (Fig. 3C; Supplementary Table S10).
We selected the most differentially expressed genes to represent a T cell–high/macrophage-low gene expression signature. Hierarchical clustering with this 18-gene signature divided the patients into two groups (Fig. 3D). The patients in the high expression cluster had a better prognosis than patients in the low expression cluster (5-year OS: 68.7% vs. 58.6%, P = 0.009; Fig. 3E). Thus, the prognostic impact of the gene signature on survival was concordant with that of the T cell–high/macrophage-low LME phenotype it represented.
To validate the prognostic impact of the T cell–high/macrophage-low LME gene signature, we used the signature to cluster the Chapuy and colleagues (7) data (n = 137; Fig. 3F; Supplementary Table S11). The samples clustered into three subgroups. Patients belonging to the high gene expression group had a better prognosis compared with the patients belonging to the low/intermediate expression group (5-year OS: 90.6% vs. 65.3%, P = 0.021; 5-year PFS: 79.9% vs. 59.5%, P = 0.038; Fig. 3G; Supplementary Fig. S10C), thus validating our findings.
Immune checkpoint–expressing macrophages have adverse impact on survival
To further understand the adverse prognostic impact of macrophages on survival, we analyzed the contribution of immunophenotypes and immune checkpoint protein expression on macrophage-associated outcome. We discovered that a high proportion of CD163−PD-L1+/TIM3+ macrophages translated to poor outcome [OS: HR = 2.60, 95% confidence interval (CI), 1.47–4.56, Padj = 0.014; PFS: HR = 2.36, 95% CI, 1.37–4.06, Padj = 0.028; Fig. 4A and B; Supplementary Fig. S11A–S11D). The survival was independent of IPI, molecular subtype, and DPE status (Fig. 4C–E; Supplementary Fig. S11E–S11G). The adverse prognostic impact of the immune checkpoint–expressing macrophages was dependent on the proportion of T cells, since the prognostic impact of PD-L1+TIM3+CD163− M1-like macrophages was evident in the patients with T cell–high, but not in patients with T cell–low LME (Fig. 4F and G; Supplementary Fig. S11H–S11I). When double-hit lymphomas were excluded from the survival analyses, prognostic impact of immune checkpoint–expressing macrophages on survival remained unchanged (Supplementary Fig. S12).
Figure 4.

Clinical impact of immune checkpoint–expressing macrophages in the DLBCL microenvironment. A, A forest plot visualizing the impact of immune checkpoint–expressing macrophages on OS in the mIHC cohort, as evaluated by Cox regression univariate analyses with continuous variables. B, Kaplan–Meier (log-rank test) survival plots depict OS in patients with high and low proportion of PD-L1+TIM3+CD163− macrophages using median cutoff in the mIHC cohort. C–E, Forest plots visualizing the impact of PD-L1+TIM3+CD163− macrophages on OS in Cox regression multivariate analyses with IPI (C), IPI and molecular subtype (D), and IPI, molecular subtype and double protein expression (E) in the mIHC cohort. F and G, Forest plots visualizing the impact of immune checkpoint–expressing macrophages on OS in the T cell–high (F) and low (G) groups, as evaluated by Cox regression univariate analyses with continuous variables in the mIHC cohort. H, Forest plot visualizing the impact of the gene expression of immune checkpoint molecules in macrophages/monocytes, inferred with CIBERSORTx, on OS in the T cell–high group in the Reddy and colleagues' cohort, as evaluated by Cox regression univariate analyses with continuous variables. *, P ≤ 0.05 after FDR correction.
Next, we utilized CIBERSORTx to study whether the expression of immune checkpoint molecule genes in macrophages associated with survival in the Reddy and colleagues (5) validation cohort. Consistent with the mIHC cohort, high expression of CD274 (PD-L1) and PDCD1LG2 (PD-L2) in macrophages correlated with poor overall survival in patients with T cell high LME (HR = 1.32, 95% CI, 1.04–1.69, P = 0.024 and HR = 2.06, 95% CI, 1.04–4.08, P = 0.038, respectively). A trend was also seen for the HAVCR2 (TIM3) expressing macrophages (HR = 2.38, 95% CI, 0.88–6.41, P = 0.087; Fig. 4H). In contrast, we did not see correlation between checkpoint molecule–expressing macrophages and outcome in the patients with T cell–low LME (Supplementary Fig. S11J).
Immune cells interacting with each other express immune checkpoint molecules
As the adverse impact of immune checkpoint–expressing macrophages on survival was dependent on the T-cell content of the tumor, we speculated that the negative signal is translated through the inhibition of T cells. Therefore, we studied the spatial relations between the immune cells in the mIHC cohort and calculated the number of their interactions (Fig. 5A). First, there were more interactions between B cells and macrophages (Padj = 0.003), between T cells with themselves (Padj = 0.002), and between T cells and macrophages (Padj = 0.017) in HLA-DR–positive lymphomas than HLA-DR–negative lymphomas (Fig. 5B). Interestingly, PD-1 and PD-L1 expression was enriched on macrophages that interacted with T cells (Padj < 0.001; Fig. 5C), suggesting that immune checkpoint molecules are involved in mediating the negative signal between macrophages and T cells. Moreover, PD-L1 and TIM3 expression, as well as PD-1, PD-L1, and TIM3 expression was enriched on macrophages that interacted with lymphoma cells and other macrophages, respectively (Padj < 0.001 for all; Fig. 5C).
Figure 5.

Interactions between different immune cell subtypes in the DLBCL microenvironment. A, The Euclidean distance d between two cells. B, Boxplots depicting differences in the number of interactions between B cells and macrophages, T cells with other T cells, as well as T cells and macrophages in HLA-DR–positive (pos), moderate (mod), and negative (neg) DLBCLs. C, Bubble plot depicting the expression of immune checkpoint molecules on macrophages that interact with other immune cells compared with macrophages that do not interact with these cells, as well as on T cells and B cells that interact with macrophages compared to T cells and B cells that do not interact with macrophages. The size of the bubbles is proportional to −log10(FDR-adjusted P value), and the color of the bubbles resembles log10FC of interacting versus noninteracting cells. D, Bubble plot depicting differences in the expression of immune checkpoint molecules on immune cells that interact (TRUE) and do not interact (FALSE) with other immune cell subtypes between HLA-ABC–positive and moderate/negative DLBCLs. The size of the bubbles is proportional to −log10(FDR-adjusted P value), and the color of the bubbles resembles log10FC of HLA-ABC–positive versus moderate/negative.
Finally, we investigated whether the expression of immune checkpoint molecules on immune cells differed between the biological subgroups. HLA-ABC positivity associated with PD-L1 and TIM3 expression on both macrophages and lymphoma cells when these cells interacted with each other. PD-L1 expression was also enriched on both macrophages and T cells when they interacted with each other in HLA-ABC–positive cases. In addition, immune checkpoint molecule expression was increased on the immune cells when they interacted with other cells displaying the same phenotype (Fig. 5D).
Discussion
Recently, the molecular and genetic landscape of DLBCL has been thoroughly characterized (5–7). In addition, classification based on the DLBCL microenvironment is emerging, and for example four distinct LME gene expression signatures, titled “GC-like”, “mesenchymal” (MS), “inflammatory” (IN), and “depleted” (DP) have been identified (14). Based on the number of tumor-infiltrating immune cells, lymphomas can also be divided into inflamed and noninflamed phenotypes, with DLBCL usually grouped as a noninflamed lymphoma, where the microenvironment has little biological and clinical significance (19). In this study, we characterized DLBCLs according to tumor-infiltrating immune cells and their interactions, and could classify the patients into clinically meaningful subgroups with differential impact on outcome. Our results indicate that DLBCLs exhibit both inflamed and noninflamed LMEs. Furthermore, the results show that while the division, based on the proportion of T cells, is sufficient to separate the two LME subgroups, the DLBCLs with inflamed LME have also higher proportions of other immune cell subtypes.
Previous studies addressing the clinical impact of the LME have mostly focused on analyzing the association of single-cell subtypes with survival with conflicting findings (13, 46–49). Our work represents a comprehensive analysis of the DLBCL LME integrating immunophenotyping and gene expression profiling with clinical characteristics and survival. In addition of studying lymphoma-infiltrating macrophage, T-cell, and NK-cell proportions separately, we addressed whether their relationships affected the outcome. Our results indicate that the amount of distinct immune cells in relation to each other in the LME has more clinical impact than the amount of these immune cells alone. While we found neither T-cell, macrophage, nor NK-cell proportions as such to associate with outcome, we could identify a group of patients with T cell–high/macrophage-low LME to have a superior outcome compared with the patients with a T cell–high/macrophage-high or T cell–low LME. Thus, it appears that macrophages can neutralize the favorable prognostic impact of T cells on survival.
In our previous study, we showed that high proportion of immune checkpoint–positive CD3−CD4+ cells translated to poor outcome, and speculated that these cells represented immune checkpoint–expressing macrophages (13). In this work, high proportion of immune checkpoint–expressing macrophages, and immune checkpoint–expressing M1-like macrophages in particular, translated to poor outcome. It is, therefore, possible that checkpoint-expressing M1-like macrophages represent the cells identified in our previous study. We also found that the adverse impact of immune checkpoint–expressing macrophages on survival was dependent on high proportions of T cells in the LME. We speculate that the negative impact of macrophages is mediated through the inhibition of T cells by immune checkpoint molecules. This theory is further strengthened by the finding that the expression of immune checkpoint molecules is enriched on macrophages that interact with T cells. Similar findings have also been seen in cHL (50).
To date, the response rates to CBT have been low in patients with DLBCL (35). This may be partially due to low incidence of genetic alterations leading to overexpression of PD-L1, which is commonly seen in cHL where CBT has shown promising efficacy (29, 51). Another possible explanation is the upregulation of other immune checkpoint molecules than PD-L1, and in such cases combination CBT may lead to better response rates. As some patients with DLBCL benefit from CBT, better patient selection may improve the efficacy of these therapies. Upregulation of immune checkpoint molecules is mainly seen in inflamed lymphomas (27, 28). Therefore, inflamed DLBCLs can be expected to respond better to CBT than noninflamed DLBCLs. The patients with high expression of the targeted immune checkpoint molecules may show better response rates as well. Furthermore, we speculate that especially the patients with inflamed DLBCLs and high proportion of macrophages may respond to CBT.
Our study has some limitations. First, as we performed mIHC analyses on TMAs, we have only studied a small fraction of lymphoma tissue in each case. It would be interesting to study whether there is intratumoral or intertumoral heterogeneity within the DLBCL microenvironment. Second, we validated our data using deconvolution analysis, and although robust and well validated, it is not equal to mIHC-based cell identification. Third, we have not studied the interactions between B cells and T cells, which may be central in explaining the positive impact T cells have in the absence of macrophages. Finally, the low number of NK cells identified in our analyses may at least partly be due to technical issues.
In conclusion, our results show that the interplay between different immune cells in the LME is clinically more meaningful than the proportions of single-cell subtypes alone. In particular, the macrophage content seems to impair the antitumoral activity of T cells, possibly by immune checkpoint–related mechanisms. However, functional experiments showing that macrophages inhibit the activity of T cells are warranted. Furthermore, our results suggest that CBT should be investigated in clinical trials in the patients with inflamed DLBCLs and high proportions of immune checkpoint–expressing immune cells.
Authors' Disclosures
O. Brück reports personal fees from Novartis, Amgen, and Sanofi outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
We thank Annabrita Schoonenberg (FIMM) for performing the mIHC stainings and the Digital and Molecular Pathology Unit supported by Helsinki University and Biocenter Finland for the mIHC stainings. Anne Aarnio is acknowledged for technical assistance. This research was funded by the grants from the Academy of Finland (to S. Leppä), Finnish Cancer Organizations (to S. Leppä), Sigrid Juselius Foundation (to S. Leppä), University of Helsinki (to S. Leppä), Helsinki University Hospital (to S. Leppä), Biomedicum Helsinki Foundation (to M. Autio), and Finska Läkaresällskapet (to M. Autio).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
M. Autio: Conceptualization, formal analysis, investigation, visualization, writing–original draft. S.-K. Leivonen: Conceptualization, formal analysis, supervision, investigation, visualization, methodology, writing–review and editing. O. Brück: Software, formal analysis, investigation, visualization, methodology, writing–review and editing. M.-L. Karjalainen-Lindsberg: Resources, methodology, writing–review and editing. T. Pellinen: Resources, formal analysis, methodology, writing–review and editing. S. Leppä: Conceptualization, resources, supervision, funding acquisition, project administration, writing–review and editing.
References
- 1. Sehn LH, Salles G. Diffuse Large B-cell lymphoma. N Engl J Med 2021;384:842–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 3. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47. [DOI] [PubMed] [Google Scholar]
- 4. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008;359:2313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell 2017;171:481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378:1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized phase III trial of Ibrutinib and Rituximab Plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol 2019;37:1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies A, Cummin TE, Barrans S, Maishman T, Mamot C, Novak U, et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): an open-label, randomised, phase 3 trial. Lancet Oncol 2019;20:649–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keane C, Gill D, Vari F, Cross D, Griffiths L, Gandhi M. CD4(+) tumor infiltrating lymphocytes are prognostic and independent of R-IPI in patients with DLBCL receiving R-CHOP chemo-immunotherapy. Am J Hematol 2013;88:273–6. [DOI] [PubMed] [Google Scholar]
- 11. Keane C, Vari F, Hertzberg M, Cao KA, Green MR, Han E, et al. Ratios of T-cell immune effectors and checkpoint molecules as prognostic biomarkers in diffuse large B-cell lymphoma: a population-based study. Lancet Haematol 2015;2:e445–55. [DOI] [PubMed] [Google Scholar]
- 12. Xu-Monette ZY, Xiao M, Au Q, Padmanabhan R, Xu B, Hoe N, et al. Immune profiling and quantitative analysis decipher the clinical role of immune-checkpoint expression in the tumor immune microenvironment of DLBCL. Cancer Immunol Res 2019;7:644–57. [DOI] [PubMed] [Google Scholar]
- 13. Autio M, Leivonen SK, Brück O, Mustjoki S, Jørgensen JM, Karjalainen-Lindsberg ML, et al. Immune cell constitution in the tumor microenvironment predicts the outcome in diffuse large B-cell lymphoma. Haematologica 2021;106:718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kotlov N, Bagaev A, Revuelta MV, Phillip JM, Cacciapuoti MT, Antysheva Z, et al. Clinical and biological subtypes of B-cell lymphoma revealed by microenvironmental signatures. Cancer Discov 2021;11:1468–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steen CB, Luca BA, Esfahani MS, Azizi A, Sworder BJ, Nabet BY, et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell 2021;39:1422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer 2014;14:517–34. [DOI] [PubMed] [Google Scholar]
- 17. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kline J, Godfrey J, Ansell SM. The immune landscape and response to immune checkpoint blockade therapy in lymphoma. Blood 2020;135:523–33. [DOI] [PubMed] [Google Scholar]
- 20. Liu WR, Shipp MA. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood 2017;130:2265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, et al. NFkappaB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood 2005;106:1392–9. [DOI] [PubMed] [Google Scholar]
- 22. Booman M, Douwes J, Glas AM, Riemersma SA, Jordanova ES, Kok K, et al. Mechanisms and effects of loss of human leukocyte antigen class II expression in immune-privileged site-associated B-cell lymphoma. Clin Cancer Res 2006;12:2698–705. [DOI] [PubMed] [Google Scholar]
- 23. Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, et al. Combined genetic inactivation of beta2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 2011;20:728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 2004;103:4251–8. [DOI] [PubMed] [Google Scholar]
- 25. Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011;471:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med 2010;362:875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010;116:3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Georgiou K, Chen L, Berglund M, Ren W, de Miranda NF, Lisboa S, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood 2016;127:3026–34. [DOI] [PubMed] [Google Scholar]
- 29. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015;372:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zinzani PL, Ribrag V, Moskowitz CH, Michot JM, Kuruvilla J, Balakumaran A, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol 2019;37:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005;105:1851–61. [DOI] [PubMed] [Google Scholar]
- 33. Godfrey J, Tumuluru S, Bao R, Leukam M, Venkataraman G, Phillip J, et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T-cell-inflamed phenotype. Blood 2019;133:2279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov 2019;9:546–63. [DOI] [PubMed] [Google Scholar]
- 35. Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, et al. Nivolumab for Relapsed/Refractory diffuse Large B-cell lymphoma in patients ineligible for or having failed autologous transplantation: A Single-Arm, Phase II Study. J Clin Oncol 2019;37:481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blom S, Paavolainen L, Bychkov D, Turkki R, Maki-Teeri P, Hemmes A, et al. Systems pathology by multiplexed immunohistochemistry and whole-slide digital image analysis. Sci Rep 2017;7:15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, et al. ilastik: interactive machine learning for (bio)image analysis. Nat Methods 2019;16:1226–32. [DOI] [PubMed] [Google Scholar]
- 38. Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006;7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meriranta L, Pasanen A, Alkodsi A, Haukka J, Karjalainen-Lindsberg ML, Leppä S. Molecular background delineates outcome of double protein expressor diffuse large B-cell lymphoma. Blood Adv 2020;4:3742–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brück O, Lee MH, Turkki R, Uski I, Penttilä P, Paavolainen L, et al. Spatial immunoprofiling of the intratumoral and peritumoral tissue of renal cell carcinoma patients. Mod Pathol 2021;34:2229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liarski VM, Sibley A, van Panhuys N, Ai J, Chang A, Kennedy D, et al. Quantifying in situ adaptive immune cell cognate interactions in humans. Nat Immunol 2019;20:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cai QC, Liao H, Lin SX, Xia Y, Wang XX, Gao Y, et al. High expression of tumor-infiltrating macrophages correlates with poor prognosis in patients with diffuse large B-cell lymphoma. Med Oncol 2012;29:2317–22. [DOI] [PubMed] [Google Scholar]
- 47. Riihijärvi S, Fiskvik I, Taskinen M, Vajavaara H, Tikkala M, Yri O, et al. Prognostic influence of macrophages in patients with diffuse large B-cell lymphoma: a correlative study from a Nordic phase II trial. Haematologica 2015;100:238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCord R, Bolen CR, Koeppen H, Kadel EE III, Oestergaard MZ, Nielsen T, et al. PD-L1 and tumor-associated macrophages in de novo DLBCL. Blood Adv 2019;3:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li L, Sun R, Miao Y, Tran T, Adams L, Roscoe N, et al. PD-1/PD-L1 expression and interaction by automated quantitative immunofluorescent analysis show adverse prognostic impact in patients with diffuse large B-cell lymphoma having T-cell infiltration: a study from the International DLBCL Consortium Program. Mod Pathol 2019;32:741–54. [DOI] [PubMed] [Google Scholar]
- 50. Carey CD, Gusenleitner D, Lipschitz M, Roemer MGM, Stack EC, Gjini E, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017;130:2420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for Relapsed/Refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.