

Abstract
Organometallic compounds offer broad scope for the design of therapeutic agents, but this avenue has yet to be widely explored. A key concept in the design of anticancer complexes is optimization of chemical reactivity to allow facile attack on the target site (e.g., DNA) yet avoid attack on other sites associated with unwanted side effects. Here, we consider how this result can be achieved for monofunctional “piano-stool” ruthenium(II) arene complexes of the type [(η6-arene)Ru(ethylenediamine)(X)]n+. A potentially important activation mechanism for reactions with biomolecules is hydrolysis. Density functional calculations suggested that aquation (substitution of X by H2O) occurs by means of a concerted ligand interchange mechanism. We studied the kinetics and equilibria for hydrolysis of 21 complexes, containing, as X, halides and pseudohalides, pyridine (py) derivatives, and a thiolate, together with benzene (bz) or a substituted bz as arene, using UV-visible spectroscopy, HPLC, and electrospray MS. The x-ray structures of six complexes are reported. In general, complexes that hydrolyze either rapidly {e.g., X = halide [arene = hexamethylbenzene (hmb)]} or moderately slowly [e.g., X = azide, dichloropyridine (arene = hmb)] are active toward A2780 human ovarian cancer cells, whereas complexes that do not aquate (e.g., X = py) are inactive. An intriguing exception is the X = thiophenolate complex, which undergoes little hydrolysis and appears to be activated by a different mechanism. The ability to tune the chemical reactivity of this class of organometallic ruthenium arene compounds should be useful in optimizing their design as anticancer agents.
Keywords: anticancer, bioorganometallic, hydrolysis, kinetics, ruthenium complexes
Organometallic chemistry has evolved rapidly during the last 50 years, notably in areas related to catalysis and materials (1). Applications in biology and medicine are in their infancy, but the potential for exciting developments is clear (2). In the field of cancer chemotherapy, the cyclopentadienyl complex [Cp2TiCl2] has been in clinical trials (3, 4), and a ferrocene derivative of Tamoxifen is a candidate for trials for breast cancer therapy (5). The successful design of second- and third-generation platinum anticancer drugs, now widely used in the clinic, has demonstrated that detailed knowledge of the factors that control ligand substitution and redox reactions is very valuable in drug design. The chemical reactivity of the complexes can be chosen so as to balance the inertness required for the drug to reach its target site (e.g., DNA) and minimize attack on other sites (side effects) yet allow activation necessary for binding to the target. Thus, cis-[PtCl2(NH3)2], cisplatin, is relatively unreactive in high-chloride media (e.g., blood plasma) and is activated by hydrolysis near DNA in the nucleus (6). In contrast, carboplatin and oxaliplatin are relatively inert to hydrolysis, have a milder spectrum of side effects, and probably attack DNA by means of chelate ring-opening reactions (7, 8). Exploration of the factors that control the aqueous chemistry of organometallic complexes may therefore also allow the design of effective anticancer agents.
Our studies are focused on monofunctional ruthenium(II) arene anticancer complexes of the type [(η6-arene)Ru(ethylenediamine)(X)]n+, where X is a leaving group (e.g., Cl). In these pseudooctahedral “piano-stool” RuII complexes, a π-bonded arene (the “seat” of the stool) occupies three coordination sites, and the two nitrogens of ethylenediamine (en) and X fill the remaining three sites (the “legs”). Although ruthenium has a rich redox chemistry (9), the presence of an arene greatly stabilizes RuII compared with RuIII (ref. 10 and references therein). These complexes can exhibit cytotoxicity toward cancer cells, including cisplatin-resistant cells (11, 12). In nucleotide adducts, {(η6-arene)Ru(en)}2+ exhibits a high selectivity for N7 of guanine and, in contrast to cisplatin, shows little interaction with adenine (13, 14). Reactions of these chloro RuII arene complexes with nucleotides appear to involve initial aquation, and Ru–OH2 bonds appear to be more reactive than Ru–OH bonds (15), a behavior parallel to that of PtII diam(m)ine anticancer complexes (16).
Here, we have studied the effect of both X and the arene on the kinetics and thermodynamics of hydrolysis of [(η6-arene)Ru(en)(X)]n+ complexes, with X ranging from halide, pseudohalide, and pyridine (py) derivatives to thiolate and with the arene ranging from benzene (bz) to more sterically hindered p-cymene (p-cym) and hexamethylbenzene (hmb) to multiring [indan (ind) and biphenyl (bip)] complexes. In view of the high chloride concentrations in blood plasma and cell culture media, we have also studied reactions with chloride and with guanine [as guanosine 5′ monophosphate (GMP)] as a potential target site on DNA. Density functional computational methods have been used to elucidate the mechanism of the aquation reactions, HPLC and electrospray ionization MS (ESI-MS) have been used to identify the products, and UV-visible (UV-Vis) spectroscopy has been used to determine rates and equilibria. The x-ray crystal structures of six complexes are reported. Although it is sometimes stated (17) that both RuII and RuIII oxidation states are “substitution-inert,” we show that the rates of hydrolysis of these RuII arene complexes can vary over many orders of magnitude, depending on the nature of X. As for square-planar PtII complexes, it is possible to design cytotoxic RuII arene complexes with widely contrasting ligand substitution rates.
Materials and Methods
Materials. The synthesis and characterization of the ruthenium arene complexes studied here, [(η6-arene)Ru(en)X][PF6]n, arene = hmb, X = Cl (1), Br(5), I(9), N3 (14), and thiophenolate (SPh) (21), n = 1; X = py (16), 3,5-dichloropyridine (dcp) (17), 3,5-difluoropyridine (dfp) (18), p-cyanopyridine (pcp) (19), and 3-picoline (pic) (20), n = 2; arene = bip, X = Cl (2), Br(6), I(10), and N3 (15), n = 1; arene = ind, X = Cl (3), Br (7), and I (11); arene = bz, X = Cl (4), Br (8), I (12); arene = p-cym, X = I (13), n = 1, are described in Supporting Text, which is published as supporting information on the PNAS web site. CF3COOH (TFAH) was purchased from Acros (Geel, Belgium), and GMP disodium salt hydrate was purchased from Aldrich.
UV-Vis Spectroscopy. A PerkinElmer Lambda-16 UV-Vis spectrophotometer was used with 1-cm path-length quartz cuvettes (0.5 ml) and a PTP1 Peltier temperature controller. Spectra were processed by using uvwinlab software for Windows 95.
HPLC and ESI-MS. The procedures were similar to those we reported in ref. 18 and are described in Supporting Text.
X-Ray Crystallography. Details of the structure determinations of complexes 1, 3, 5, 9, 11, and 19 are provided in Tables 4–7 and Figs. 3–8, which are published as supporting information on the PNAS web site.
Hydrolysis Equilibria. Normalized areas of HPLC peaks detected by UV absorption at wavelengths where the absorption of aqua complexes and their respective parent species presented minimum differences were used to calculate the hydrolysis equilibrium constants (K) according to Eq. 1:
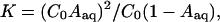 |
[1] |
where C0 is the initial concentration of the parent complex, and Aaq is the normalized area of the HPLC peak for the aqua complex.
Kinetics. Details of the procedures used to determine rate constants for substitution of X by H2O, Cl, or GMP by UV-Vis spectroscopy are given in Supporting Text.
IC50 Values. The concentrations of the complexes that caused 50% inhibition of the growth of human ovarian A2780 cancer cells were determined as described in the Supporting Text.
Partition Coefficients and Cell/DNA Uptake. The octanol/water partition coefficients and uptake of complexes 1 and 21 by A2780 human ovarian cancer cells and the extent of ruthenation of their DNA were determined as described in Supporting Text.
Computation. Density functional theory calculations were performed by using the amsterdam density functional (adf) (19) program suite (version 2003.01) (20). Geometries and energies were obtained by using the nonrelativistic Perdew/Wang gradient-corrected functional (PW91) unless otherwise stated. The frozen-core approximation (21) was applied with the orbitals up to and including that labeled in brackets as follows: Ru[3d], O[1s], C[1s], N[1s], Cl[2p], Br[4p], and I[4d]. Basis sets and other details are described in Supporting Text.
Supporting Information. Details of experimental procedures (syntheses; HPLC; ESI-MS; x-ray crystallography; computations; kinetics; partition coefficients; cell/DNA uptake; crystallographic, computational, and kinetic data; and separation and identification of hydrolysis and GMP products) are provided in Tables 4–10 and Figs. 3–26, which are published as supporting information on the PNAS web site.
Results
First, we attempted to gain insight into the potential role of the ligand X in the mechanism of aquation of [(η6-arene)Ru(en)X]n+ complexes by using computational methods.
Computation. A test of the structural accuracy of the functional PW91 was performed by comparing the fully optimized structure of [(η6-bz)Ru(en)Cl]+ with the x-ray crystal structure of [(η6-p-cym)Ru(en)Cl][PF6] (12). PW91 overestimated the Ru–ligand contacts by ≈0.03–0.04 Å, except the Ru–Cl distance (2.40 Å), which was shorter than experiment (2.44 Å). Overall, the agreement with experimental data was satisfactory.
The reaction modeled was {[(η6-bz)Ru(en)X]+·H2O} → [TS] → {[(η6-bz)Ru(en)(H2O)]2+·X–}, X = Cl, Br, I, and N3 (where TS is transition state).
The system retained the entering and leaving groups within the second coordination sphere as appropriate. This substitution process can be described in terms of the two parameters Ru–L and Ru–OH2. In principle, the process could be associative or dissociative, so a preliminary set of calculations was undertaken to map out the potential energy surface as a function of these two variables. For X = Cl, the reaction potential energy surface is shown in Fig. 1. An approximate saddle point is located at Ru–O ≈ 2.8 Å and Ru–Cl ≈3.0 Å, and the corresponding structure was used in a full transition state optimization leading to a structure with Ru–O = 2.99 Å and Ru–Cl = 3.28 Å lying 17 kcal·mol–1 above the reactants.
Fig. 1.

Reaction potential energy surface for the hydrolysis of [(η6-bz)Ru(en)Cl]+.
The barrier height does not vary dramatically with different functionals (Table 8), nor does it change when the arene ligand is changed from simple bz to hmb. Having established that PW91/ basis I with bz as the arene ligand is a suitable model system, we then explored the effect of varying X, assuming that the same interchange mechanism applies. The results show that the reaction barriers and overall reaction energies for the aquation of the halide (Cl, Br, and I) and pseudohalide N3 in {(η6-bz)Ru(en)}2+ complexes follow the order Br < Cl < I < N3 (Table 1).
Table 1. Selected bond lengths, forward reaction barriers, and overall reaction energies from density functional theory calculations for reactions {[(η6-bz)Ru(en)X]n+·H2O} → [TS] → {[(η6-bz)Ru(en)(H2O)]2+·X}.
| X | Ru-X/Ru-OH2 reactant, Å | Ru-X/Ru-OH2 TS, Å | Ru-X/Ru-OH2 product, Å | ΔE‡, kcal·mol-1 | ΔEreac, kcal·mol-1* |
|---|---|---|---|---|---|
| Cl | 2.47/3.94 | 3.28/2.99 | 4.10/2.20 | 17.0 | 2.5 |
| Br | 2.63/3.99 | 3.39/2.88 | 4.24/2.21 | 16.0 | 1.9 |
| I | 2.85/4.44 | 3.66/2.95 | 4.48/2.22 | 18.1 | 5.2 |
| N3 | 2.15/4.34 | 2.77/3.00 | 3.75/2.19 | 25.4 | 8.2 |
| pcp | 2.11/3.89 | 2.95/2.75 | 3.92/2.18 | 8.4 | 6.7 |
| dcp | 2.13/4.04 | 2.90/2.67 | 3.92/2.17 | 21.6 | 5.2 |
| dfp | 2.12/4.08 | 2.89/2.71 | 4.00/2.18 | 22.6 | 5.4 |
| py | 2.12/3.85 | 2.91/2.77 | 3.88/2.17 | 23.5 | 5.1 |
| pic | 2.12/3.73 | 2.84/2.72 | 3.88/2.17 | 22.6 | 3.9 |
TS, transition state.
ΔEreac values relative to reactant species at zero.
Next, the anionic X ligand was replaced by a neutral py donor, either py itself or pcp, dcp, dfp, or pic. A similar concerted mechanism was used with entering and leaving groups remaining in the second coordination sphere. The calculated order of hydrolysis rates based on the forward reaction barrier heights is pcp >> dcp > dfp = pic > py, although it should be noted that the barriers for the last four ligands span only ≈2 kcal·mol–1 (Table 1).
These data suggested that the nature of X could control the hydrolysis of these complexes, and this hypothesis was confirmed by the following experiments.
Synthesis and Characterization of Complexes. The syntheses followed previously described routes (12, 22–24). The x-ray crystal structures of complexes 2, 13, 22, and 23 have already been reported (12, 13); those of the halo complexes [(η6-hmb)Ru(en)X][PF6], X = Cl (1), Br (5), I (9), [(η6-ind)Ru(en)X][PF6](X = Cl (3), and I (11), as well as the pcp complex [(η6-hmb)Ru(en)(pcp)][PF6]2 (19), are reported here (details are provided in Supporting Text). All adopt the familiar piano-stool structure (Figs. 3–8).
Hydrolysis Products. Aqueous solutions of [(η6-hmb)Ru(en) X][PF6]n [X = Cl (1), Br(5), I(9), N3 (14), dcp (17), dfp (18), pcp (19), and pic (20)] were allowed to equilibrate for 24–48 h at ambient temperature and were then analyzed by HPLC. Two well separated peaks were observed for each complex corresponding to the aqua adduct and the respective parent cation (Fig. 9), as identified by the subsequent ESI-MS assays (Figs. 10 and 11). As an example, for complex 9 [(η6-hmb)Ru(en)I][PF6], the fraction eluting at 3.76 min gave rise to two ion peaks at m/z 323.0 and 437.1, assignable on the basis of mass-to-charge ratios and isotopic models (18) (Fig. 10) to the aqua complex [(η6-hmb)Ru(en)(H2O)]2+ (calculated m/z of 323.1 for [(η6-hmb)Ru(en)(H2O)]2+–H2O–H+) and its trifluoroacetato (TFA) adduct (calculated m/z of 437.1 for [(η6-hmb)Ru(en)(TFA)]+), respectively. The fraction eluting at 11.32 min gave a peak at m/z = 450.8 corresponding to the intact cation of 9 (calculated m/z of 451.0 for 9+). Analogous products in aqueous equilibrium solutions of complexes 1, 3, 12, 13, 14, 15, 17, 18, 19, and 20 were also identified by ESI-MS analysis of their HPLC fractions (Figs. 11–15).
Hydrolysis Kinetics. Dissolution of compounds 1–21 in 19:1 mixtures of water and methanol at 298 K gave rise to hydrolysis as indicated by the concomitant changes in UV-Vis absorption bands. A typical time evolution for [(η6-ind)Ru(en)I][PF6](11) at 298 K is shown in Fig. 16. The first-order exponential decay of the absorbance of 11 at 270 nm gave a hydrolysis rate constant kH2O of 5.58 × 10–4 s–1. The hydrolysis rate constants and half-lives for compounds 1–21 are shown in Fig. 2 and listed in Tables 2, 9, and 10.
Fig. 2.

Correlation of hydrolysis with cytotoxicity. (Upper) Structures of complexes, arenes, and leaving groups. (Lower) Hydrolysis rates, equilibrium percentage of total Ru as [(η6-hmb)Ru(en)(H2O)]2+ ([Ru(H2O)]e %), and A2780 human ovarian cancer cell IC50 values for [(η6-hmb)Ru(en)X]n+ complexes with various leaving groups (X). Data for other arene complexes are listed in Table 2.
Table 2. Hydrolysis data for [(η6-arene)Ru(en)X][PF6]n (n = 1 or 2) complexes at 298 K and cytotoxicity (IC50) toward A2780 human ovarian cancer cells.
| Complex | Arene | X | kH2O, 10-3 s-1 | [Ru(H2O)]e, % | IC50, μM |
|---|---|---|---|---|---|
| 18 | hmb | dfp | 0.0208 ± 0.0001 | 32.6 | ND |
| 2 | bip | Cl | 1.24 ± 0.13 | 76.2 | 5† |
| 6 | bip | Br | 1.05 ± 0.04 | ND | ND |
| 10 | bip | I | 0.321 ± 0.045 | ND | 5 |
| 15 | bip | N3 | 0.0315 ± 0.0043 | 4.5 | 4 |
| 3 | ind | Cl | 2.29 ± 0.14 | 76.3 | 8 |
| 7 | ind | Br | 2.16 ± 0.05 | ND | ND |
| 11 | ind | I | 0.512 ± 0.046 | ND | ND |
| 4 | bz | Cl | 1.98 ± 0.02 | 98.1 | 17† |
| 8 | bz | Br | 1.50 ± 0.02 | ND | ND |
| 12 | bz | I | 0.294 ± 0.003 | 20.5 | 20† |
| 13 | p-cym | I | 0.948 ± 0.008 | 5.0 | 9† |
| 22 | dha | Cl | 2.23 ± 0.02 | 80.4* | 2† |
| 23 | tha | Cl | 2.36 ± 0.02 | 80.0* | 0.5† |
The hydrolysis rates vary over several orders of magnitude. Within each group of complexes containing the same coordinated arene, the hydrolysis rate decreases in the order Cl ≈ Br > I. Replacement of chloride (in complex 1 or 2) by the pseudohalide N3 (to give complexes 14 and 15) has an even greater effect on the hydrolysis rate (Fig. 2 and Table 2), slowing it down by ≈40-fold. Changing the leaving group to py, pic, or SPh, as in 16, 20, and 21, slows down hydrolysis dramatically, such that it was too slow to observe. The introduction of a substituted py, however, such as dcp in 17 and dfp in 18, led to observable hydrolysis, albeit slower than the corresponding Cl complex by a factor of ≈1,400. The hydrolysis rate of the pcp complex 19 was similar to that of the N3 complex 14.
For complexes containing the same leaving group Cl, the hydrolysis rate decreased with variation of the coordinated arene in the order hmb > tetrahydroanthracene (tha) ≈ dihydroanthracene (dha) (18) ≈ ind > bz > bip, and for I complexes the order was hmb > p-cym > ind > bip ≈ bz (Tables 2 and 9).
Hydrolysis Equilibria. The Cl and Br complexes hydrolyzed to >75% at equilibrium, but the I complexes (9, 12, and 13) were <50% hydrolyzed, the least being for the p-cym complex 13 (5%). Similarly, the azide compounds hydrolyzed only to a very small extent, <5% for complexes 14 and 15 (Fig. 2 and Table 2).
Although the dcp, dfp, and pcp complexes 17, 18, and 19 hydrolyze only slowly, the extent of their hydrolysis at equilibrium is significant (32–60%). However, no significant aquation (<1%) of the py and pic complexes 16 and 20 was detected by HPLC.
Substitution of I, N3, dcp, py, and SPh by Cl. The possibility that some complexes might be transformed into their chloro analogs in high-chloride (biological) media was investigated. At 310 K, significant changes in the UV-Vis absorption of complexes 13 (p-cym/I), 15 (bip/N3), 17 (hmb/dcp), and 21 (hmb/SPh) were observed in 104 mM NaCl solution (Figs. 17, 18, 20, and 24). HPLC separations (Figs. 17, 18, 20, and 25) indicated that the majority of the I leaving group in 13 was substituted by Cl, but N3 in 15 and dcp in 17 were only partially substituted after 24 h and for SPh < 3%. The variation of absorption with time gave rise to the reaction half-lives listed in Table 3. Both UV-Vis spectra and HPLC assays (Fig. 19) showed that no substitution of the py ligand in 16 by Cl occurred within 24 h.
Table 3. Half-lives for substitution reactions of leaving groups in [(η6-arene)Ru(en)X]n+ by H2O, Cl, and GMP at 298 K.
|
t1/2, min
|
||||
|---|---|---|---|---|
| Complex (arene) | X | H2O | Cl | GMP |
| 1 (hmb) | Cl | 0.44 | 24.7 | |
| 13 (p-cym) | I | 12.2 | 2.73 | 22.6 |
| 15 (bip) | N3 | 367 | 1122 | 554 |
| 16 (hmb) | py | — | — | — |
| 17 (hmb) | dcp | 537 | 631 | 578 |
| 21 (hmb) | SPh | — | 107 | 631 |
—, no reaction.
Partition Coefficients and Cell/DNA Uptake. The octanol/water partition coefficients (log Poct) of 1 and 21 were determined to be –1.53 ± 0.02 and –0.17 ± 0.02, respectively. Treatment of A2780 human ovarian cancer cells with 20 μM 1 or 21 for 24 h gave rise to cell/DNA-bound [Ru] levels of 74 ± 3/7.1 ± 4.0 and 136 ± 5/4.2 ± 2.7 pmol per 106 cells, respectively.
Reactions of N3, py, dcp, and SPh Complexes with GMP. The possibility that slowly aquating complexes could react directly with guanine was investigated. Reactions of 0.5 mM 15, 16, 17, or 21 with 0.5 mM GMP were followed by UV-Vis spectrometry at 298 K. The py complex 16 did not react (Fig. 22). Reaction half-lives were determined from absorbance-versus-time plots for 15, 17, and 21 and, for comparison, for 1 and 13, which undergo fast aquation (Figs. 21, 23, 24, and 26 and Table 3).
Positive-ion mass spectra of HPLC fractions (Figs. 21–23 and 25) indicated that the second fractions for reactions of 15 and 17 (identical to that for 21) with GMP contain GMP adducts, giving rise to singly charged ion peaks at m/z = 678.2 (calculated m/z of 678.1 for [(η6-bip)Ru(en)(GMP)] + H+) and 686.3 (calculated m/z of 686.2 for [(η6-hmb)Ru(en)(GMP)] + H+), respectively. No GMP adduct of 16 was detectable (Fig. 22). Hence, it appears that N3, dcp, and SPh can all be substituted by GMP.
Cytotoxicity. The concentrations of complexes that achieved 50% inhibition of the growth of human ovarian A2780 cancer cells (IC50 values) were determined (Fig. 2 and Table 2). The activity of most of the complexes against this cancer cell line is at least comparable to that of carboplatin (IC50 = 6 μM), and several approach that of cisplatin (IC50 = 0.6 μM). Exceptions are the hmb/py and hmb/pic complexes 16 and 20 (for which no significant hydrolysis was observed); the former is inactive, and the latter is weakly active. For complexes containing the same leaving group, the cytotoxicity increases with the size of the coordinated arene in the order bz < p-cym (11) < hmb < bip < dha < tha.
It is notable that the Cl and N3 complexes containing either hmb or bip as arene ligands all exhibit good activity (IC50 values of 4–18 μM) despite the significant difference in their rates and extent of hydrolysis. Also, the thiolate complex 21 exhibits significant activity despite being relatively inert to hydrolysis.
Discussion
Mechanism of Hydrolysis. The density functional theory calculations (Fig. 1) suggested that aquation of [(η6-bz)Ru(en)X]n+ complexes proceeds via a concerted interchange pathway rather than a stepwise dissociation/coordination process. The reaction does not appear to be strongly associatively nor dissociatively activated, because the Ru–Cl bond at the transition state extends by 0.81 Å relative to the reactant species, whereas the Ru–O bond is 0.79 Å longer than in the aqua product (Table 1).
In an interchange associative pathway (Ia), bond-breaking alone is not rate controlling, and a heavier halide makes crowding of the central Ru atom unfavorable in associative states with a higher coordination number (seven) (25, 26). The effect of halides on the hydrolysis rate of RuII arene complexes is opposite to that reported for platinum compounds [PtXn(H2O)4–n](2–n)+, for which Br analogs of Cl complexes hydrolyze faster in all three hydrolysis steps (27).
The effective electronegativity of N3 lies between that of Cl and Br (28); however, the hydrolysis rates of N3 complexes 14 and 15 are much lower than those of the Cl and Br analogs. This lower rate may be due to the increased steric bulk of this polyatomic pseudohalide; an Ia substitution pathway is more influenced by steric factors than an Id (dissociative) pathway (18).
For arene en RuII halide complexes, the hydrolysis rates decrease with increase in the electron-accepting ability (29) of the arene (hmb < p-cym < tha ≈ dha ≈ indan < bip < bz), with the exception that aquation of bz complexes is slightly faster than for the bip analogs, perhaps due to steric hindrance from the pendant phenyl ring being twisted toward X (12, 18). We used 19:1 H2O:MeOH unbuffered solutions for hydrolysis (as well as Cl and GMP) studies because the aqua adducts have high pKa values (≈8.0) and hydrolysis reduces the pH only slightly [from 7 to ≈6.2 for 2, 22, and 23 (18)]. The calculated reaction barriers and overall reaction energies for the aquation of the halide and azide {(η6-bz)Ru(en)}2+ complexes follow the order Br < Cl < I < N3 (Table 1), in agreement with the experimental hydrolysis rates, Cl ≈ Br < I < N3 (Fig. 1), confirming that the higher activation energies are responsible for the slower hydrolysis of the I and N3 complexes. The larger leaving groups py, pic, and SPh in 16, 20, and 21 make the Ru center less accessible to an incoming ligand. However, electron-accepting substituents on the py ring such as Cl, F, or CN, as in 17, 18, and 19, weaken the donor ability of the py nitrogen, as seen by the much lower pKa values of dfp, dcp, and pcp (<2) compared with pic and py (5–6) (30) and lead to an increase in the hydrolysis rate.
Given the different charge states, direct comparisons of calculated activation energies for negative (Cl, Br, I, and N3) and neutral (py and py derivatives) X ligands (Table 1) are problematic. For example, the hydrolysis rate of the hmb/Cl complex 1 is ≈100-fold faster than that of the hmb/pcp complex 19, but the forward reaction barrier for 19 is only half that of 1. However, among the py and py-derivative complexes, the calculated order of hydrolysis rates based on the forward reaction barrier heights (pcp >> dcp > dfp = pic > py) is in agreement with the experimental data: The hydrolysis rate of the hmb/pcp complex (19) is ≈12 times faster than that of the hmb/dcp and hmb/dfp compounds 17 and 18, and no hydrolysis was observed for the hmb/py complex 16, although the difference between the barriers for the four complexes is only ≈2 kcal·mol–1. An exception is that no hydrolysis was observed for the hmb/pic compound 20 by UV-Vis spectroscopy, although the calculated reaction barrier is the same as that for 18 (half-life of 555 min). However, the very small equilibrium constant (Table 10) and very small change in the UV-Vis absorption made the rate difficult to determine.
Within this family of (arene)RuII complexes, therefore, the hydrolysis rates are tunable, which is potentially useful in the design of anticancer complexes. Hydrolysis is known to be an important mechanism of activation for the anticancer drug cisplatin (6), which has hydrolysis rate constants of 7.56 × 10–5 and 6.32 × 10–5 s–1 for the first and second chloride ligands, respectively (31), significantly lower than the rate constants of Ru–Cl hydrolysis in these arene complexes. However, replacing Cl by pyridyl ligands can readily effect a 40- to 1,400-fold deceleration. In the case of compounds 17, 18, and 19, these alterations result in compounds with hydrolysis rates comparable to cisplatin. A variety of other py derivatives could be used for fine-tuning.
In {(η6-arene)RuII(en)}2+ complexes, the chelated en and arene ligands also influence hydrolysis rates. Our previous work (18) has shown that en cis to the leaving group (Cl) slows down aquation of RuII arene (bip, dha, and tha) complexes, just as 2,2′-bipyridine (bpy) slows down substitution of the aqua ligand in [(η6-C6H6)Ru(bpy)(H2O)]2+ (32). Replacement of en by acetylacetonate to give {(η6-arene)RuII(acac)}+ complexes accelerates hydrolysis (33), supporting the above arguments.
Arene ligands such as bz are reported to exhibit a strong translabilizing effect for the aqua ligand in [(η6-C6H6)Ru(H2O)3]2+ (34). Also, this class of strong π-acid ligands is able to accept electron density from the central RuII giving rise to a higher charge on the metal, so that RuII in {(η6-arene)RuII}2+ behaves more like aRuIII center (18, 35). Acidic hydrolysis of RuIII complexes such as [Ru(NH3)4X2]+ and [Ru(NH3)5X]2+ (X = Cl, Br, and I) occurs via an SN2 associative pathway (36) in which bond-making is more important than bond-breaking. Therefore, the electron-accepting effect of strong π-acid arene ligands might be responsible for the shift toward a more associative pathway in the Id ↔ Ia mechanistic continuum for the {(η6-arene)RuII(en)}2+ complexes studied here (36, 37).
Activation Pathways and Cytotoxicity. Our previous work has shown that the interaction of [(η6-bip)Ru(en)Cl]+ (2) with amino acids (38, 39), peptides (40), proteins (39), and DNA bases (15) involves aquation as the first step. The chloro complexes are relatively inert. Such behavior appears to parallel that of the chloro PtII anticancer complex cisplatin, the activation of which involves intracellular aquation (6, 16, 41). The aqua ligands are much more reactive, for example, toward substitution by guanine bases on DNA (42).
In general, for the arene complexes studied here, those that hydrolyze exhibit high cytotoxicity, and those that do not hydrolyze are inactive or weakly active. For optimizing drug design and reducing side effects, it is sometimes beneficial to decrease chemical reactivity but retain cytotoxicity. Therefore, our discovery of complexes that hydrolyze at only moderate rates (e.g., hmb/N3 and hmb/dcp) or extremely slowly (hmb/SPh) yet exhibit significant activity may provide valuable leads for drug optimization.
The concentration of chloride in blood plasma or culture medium is ≈104 mM, and the cytoplasmic [Cl] is ≈22 mM (43). Therefore, substitution of nonchloride leaving groups such as I, Br, and N3 by Cl in these media could be involved in the mechanism of action. In 104 mM NaCl solution, the substitution of the I leaving group in 13 was complete within minutes at 298 K, with a half-life (2.7 min) much shorter than that of the hydrolysis (12.2 min), implying that I in 13 will be substituted by Cl instead of H2O when this complex is dissolved in 104 mM NaCl solution. In contrast, under the same conditions, the half-lives for substitution of N3 in 15 (1,120 min) and dcp in 17 (631 min) by Cl are longer than those for hydrolysis of the respective complexes (367 and 537 min, respectively), indicating that the substitution of N3 and dcp by Cl involves aquation as the first step in the reaction.
About twice as much Ru from the SPh complex 21 was taken up by A2780 cells as from the chloro complex 1, in line with its (23 times) higher octanol/water partition coefficient. However, the level of ruthenation of cellular DNA was ≈2 times higher for 1, consistent with its 2.5-times higher cytotoxicity (Fig. 2 and Table 9). This finding suggests that DNA may be an important target site for RuII arene complexes, in agreement with our previous work, which showed specific binding to G bases (12, 40, 44). Therefore, in this work, we used GMP as a model to study activation pathways. For the bip/N3 (15) and hmb/dcp (17) complexes, GMP binding (t1/2 = 554 and 578 min, respectively) is faster than substitution of N3 and dcp by Cl (1,122 and 631 min) but slower than hydrolysis (367 and 537 min). Because the half-lives for reactions of the aqua adducts of bip/Cl (2) and hmb/Cl (1) with GMP are 108 (15) and 24.3 (24.7–0.44; Table 3) min, respectively, these data strongly suggest that the reactions of 15 and 17 with DNA (GMP) will involve aquation of the parent complexes as the first (rate-controlling) step. In contrast, the py complex 16 does not hydrolyze or react with Cl or GMP and is inactive toward A2780 cells.
Interestingly, the active SPh complex 21 does not undergo aquation, and substitution of SPh by Cl occurs at a rate much faster than the GMP binding (Table 3) but to a very low extent (≈3% under [Cl] = 104 mM). Therefore, 21 may react directly with guanine, especially in the cell nucleus where [Cl] is low (4 mM; ref. 45). This reaction pathway is analogous to that of the second-generation platinum drug carboplatin, which is stable in water and reacts with chloride only very slowly and directly with nucleophiles. It seems likely that 21 is activated by means of oxidation of the bound SPh to the sulfenate or sulfinate by oxygen, analogous to our recent finding for glutathione complexes (40). Glutathione sulfenate is readily displaced from Ru by N7 of guanine.
Conclusions
We have shown that ligand substitution reactions of organometallic RuII arene complexes can be finely controlled, not only by the choice of the arene but also by the other ligands, in particular by a chelated ethylenediamine and a monofunctional leaving group (X). The hydrolysis rates of [(η6-arene)Ru(en)(X)]n+ complexes vary over many orders of magnitude, from half-lives of seconds (hmb/Cl) to minutes (e.g., hmb/N3) to hours (hmb/dfp, dcp; bip/N3) or half-lives that are too slow to be measured (hmb/py, SPh) at 298 K. Density functional theory calculations suggest that aquation occurs via a more associative pathway in a Ia ↔ Id mechanistic continuum for which bond-making is of greater importance than bond-breaking. For bz complexes, the calculated reaction barriers and overall reaction energies follow the order Cl ≈ Br < I < N3, in agreement with experimental data. Hydrolysis provides a pathway for activation. In general, complexes that are readily hydrolyzed are cytotoxic (e.g., hmb/ halide), and those that do not hydrolyze (e.g., hmb/py or pic) are inactive or weakly active.
The Br and I complexes may be prodrugs for the Cl complex in view of the high concentration of Cl in extracellular biological media (≈104 mM), but py in the biologically inactive hmb complex 16 cannot readily be substituted by Cl, and the half-lives for substitution of N3 and dcp by Cl are longer than those for hydrolysis. For the latter complexes, the hydrolysis activation pathway predominates, as it does for the Cl complexes.
An unexpected exception is the moderately active hmb/SPh complex 21, which undergoes little hydrolysis or reaction with Cl yet reacts with GMP at a similar rate to the hmb/dcp complex. It seems likely that the thiolate ligand in 21 is oxidized to sulfenate or sulfinate, which provides a facile route for displacement by guanine (DNA; ref. 40).
These findings provide a potential strategy for optimizing the anticancer activity of half-sandwich Ru(II) arene complexes whilst minimizing side effects by tuning substitution reactions, a strategy that has previously been successful for Pt anticancer drugs. Wider exploration of the kinetics and thermodynamics of metal–ligand substitution reactions should aid the tuning of the biological activity of many other families of metal complexes and of organometallic complexes in particular.
Supplementary Material
Acknowledgments
We thank Emily Jones (Oncosense Ltd.) for determining the IC50 values of 16, 19, 20, and 21; Dr. David Lyon for assistance with inductively coupled plasma MS; and the European Community, the Engineering and Physical Sciences Research Council, Biotechnology and Biological Sciences Research Council (RASOR), the Edinburgh Technology Fund, Oncosense Ltd., and Cancer Research UK for their support.
Author contributions: F.W., D.I.J., and P.J.S. designed research; F.W., A.H., E.P.L.v.d.G., R.F., M.M., R.J.D., R.A., S.G., F.P.A.F., P.L.-C., I.D.H.O., and S.P. performed research; F.W., A.H., E.P.L.v.d.G., R.J.D., R.A., S.G., F.P.A.F., P.L.-C., I.D.H.O., D.I.J., S.P., and P.J.S. analyzed data; and F.W., R.J.D., S.P., and P.J.S. wrote the paper.
Conflict of interest statement: University of Edinburgh has submitted patent applications relating to the compounds used in this study for which an exclusive license has been granted to Oncosense Ltd.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: bip, biphenyl; bz, benzene; dcp, 3,5-dichloropyridine; dfp, 3,5-difluoropyridine; dha, dihydroanthracene; en, ethylenediamine; ESI-MS, electrospray ionization MS; hmb, hexamethylbenzene; ind, indan; pic, 3-picoline (3-methylpyridine); pcp, p-cyanopyridine; p-cym, p-cymene; py, pyridine; SPh, thiophenolate; tha, tetrahydroanthracene; UV-Vis, UV-visible.
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference nos. 288192–288197). The x-ray crystallographic data for complexes 1, 3, 5, 9, 11, and 19 can be found in Data Sets 1–6, which are published as supporting information on the PNAS web site.
References
- 1.Halpern, J. (2001) Pure Appl. Chem. 73, 209–220. [Google Scholar]
- 2.Fish, R. H. & Jaouen, G. (2003) Organometallics 22, 2166–2177. [Google Scholar]
- 3.Köpf-Maier, P. & Köpf, H. (1987) Chem. Rev. 87, 1137–1152. [Google Scholar]
- 4.Sadler, P. J. (1991) Adv. Inorg. Chem. 36, 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaouen, G., Top. S., Vessieres, A. & Alberto, R. (2000) J. Organomet. Chem. 600, 23–36. [Google Scholar]
- 6.Howe-Grant, M. E. & Lippard, S. J. (1980) Met. Ions Biol. Syst. 11, 63–125. [Google Scholar]
- 7.Frey, U., Ranford, J. D. & Sadler, P. J. (1993) Inorg. Chem. 32, 1333–1340. [Google Scholar]
- 8.Barnham, K. J., Djuran, M. I., Murdoch, P. d. S., Ranford, J. D. & Sadler, P. J. (1996) Inorg. Chem. 35, 1065–1072. [DOI] [PubMed] [Google Scholar]
- 9.Clarke, M. J. (2003) Coord. Chem. Rev. 236, 209–233. [Google Scholar]
- 10.Bennett, M. A., Byrnes, M. J. & Kovacik, I. (2004) J. Organomet. Chem. 689, 4463–4474. [Google Scholar]
- 11.Aird, R. E., Cummings, J., Ritchie, A. A., Muir, M., Morris, R. E., Chen, H., Sadler, P. J. & Jodrell, D. I. (2002) Br. J. Cancer 86, 1652–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris, R. E., Aird, R. E., Murdoch, P. D., Chen, H. M., Cummings, J., Hughes, N. D., Parsons, S., Parkin, A., Boyd, G., Jodrell, D. I. & Sadler, P. J. (2001) J. Med. Chem. 44, 3616–3621. [DOI] [PubMed] [Google Scholar]
- 13.Chen, H. M., Parkinson, J. A., Parsons, S., Coxall, R. A., Gould, R. O. & Sadler, P. J. (2002) J. Am. Chem. Soc. 124, 3064–3082. [DOI] [PubMed] [Google Scholar]
- 14.Chen, H. M., Parkinson, J. A., Nováková, O., Bella, J., Wang, F. Y., Dawson, A., Gould, R., Parsons, S., Brabec, V. & Sadler, P. J. (2003) Proc. Natl. Acad. Sci. USA 100, 14623–14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen, H. M., Parkinson, J. A., Morris, R. E. & Sadler, P. J. (2003) J. Am. Chem. Soc. 125, 173–186. [DOI] [PubMed] [Google Scholar]
- 16.Guo, Z. J. & Sadler, P. J. (2000) Adv. Inorg. Chem. 49, 183–306. [Google Scholar]
- 17.Tobe, M. L. & Burgess, J. (1999) Inorganic Reaction Mechanisms (Longman, New York).
- 18.Wang, F. Y., Chen, H. M., Parsons, S., Oswald, L. D. H., Davidson, J. E. & Sadler, P. J. (2003) Chem. Eur. J. 9, 5810–5820. [DOI] [PubMed] [Google Scholar]
- 19.Velde, G. T., Bickelhaupt, F. M., Baerends, E. J., Guerra, C. F., Van Gisbergen, S. J. A., Snijders, J. G. & Ziegler, T. (2001) J. Comput. Chem. 22, 931–967. [Google Scholar]
- 20.Baerends, E. J., Bérces, A., Bo, C., Boerrigter, P. M., Cavallo, L., Deng, L., Dickson, R. M., Ellis, D. E., Fan, L., Fischer, T. H., et al. (2000) ADF 2000.01 (Free University, Amsterdam).
- 21.Baerends, E. J., Ros, P. & Ellis, D. E. (1972) Theor. Chim. Acta 27, 339–354. [Google Scholar]
- 22.Bennett, M. A., Huang, T. N., Matheson, T. W. & Smith, A. K. (1982) Inorg. Synth. 21, 74–78. [Google Scholar]
- 23.Bennett, M. A. & Smith, A. K. (1974) J. Chem. Soc. Dalton Trans., 233–241.
- 24.Zelonka, R. A. & Baird, M. C. (1972) Can. J. Chem. 50, 3063–3072. [Google Scholar]
- 25.Basolo, F. & Pearson, R. G. (1967) Mechanisms of Inorganic Reactions: A Study of Metal Complexes in Solution (Wiley, New York), 2nd Ed., pp. 124–246.
- 26.Broomhead, J. A. & Kane-Maguire, L. A. P. (1968) Inorg. Chem. 7, 2519–2523. [Google Scholar]
- 27.Hindmarsh, K., House, D. A. & Turnbull, M. M. (1997) Inorg. Chim. Acta 257, 11–18. [Google Scholar]
- 28.Hollebone, B. R. (1971) J. Chem. Soc. A, 481–486.
- 29.Carey, F. A. (1987) Organic Chemistry (MacGraw–Hill, London), pp. 430–467.
- 30.Chrystiuk, E. & Williams, A. (1987) J. Am. Chem. Soc. 109, 3040–3046. [Google Scholar]
- 31.Miller, S. E. & House, D. A. (1991) Inorg. Chim. Acta 187, 125–132. [Google Scholar]
- 32.Dadci, L., Elias, H., Frey, U., Hornig, A., Koelle, U., Merbach, A. E., Paulus, H. & Schneider, J. S. (1995) Inorg. Chem. 34, 306–315. [Google Scholar]
- 33.Fernández, R., Melchart, M., Habtemariam, A., Parsons, S. & Sadler, P. J. (2004) Chem. Eur. J. 10, 5173–5179. [DOI] [PubMed] [Google Scholar]
- 34.Rapaport, I., Helm, L., Merbach, A. E., Bernhard, P. & Ludi, A. (1988) Inorg. Chem. 27, 873–879. [Google Scholar]
- 35.Steblerrothlisberger, M., Hummel, W., Pittet, P. A., Burgi, H. B., Ludi, A. & Merbach, A. E. (1988) Inorg. Chem. 27, 1358–1363. [Google Scholar]
- 36.Broomhead, J. A., Basolo, F. & Pearson, R. G. (1964) Inorg. Chem. 3, 826–832. [Google Scholar]
- 37.Bosnich, B. & Dwyer, F. P. (1966) Aust. J. Chem. 19, 2229–2233. [Google Scholar]
- 38.Wang, F. Y., Chen, H. M., Parkinson, J. A., Murdoch, P. D. & Sadler, P. J. (2002) Inorg. Chem. 41, 4509–4523. [DOI] [PubMed] [Google Scholar]
- 39.Wang, F. Y., Bella, J., Parkinson, J. A & Sadler, P. J. (2005) J. Biol. Inorg. Chem. 10, 147–155. [DOI] [PubMed] [Google Scholar]
- 40.Wang, F. Y., Xu, J. J., Habtemariam, A., Bella, J. & Sadler, P. J. (2005) J. Am. Chem. Soc., in press. [DOI] [PubMed]
- 41.Reedijk, J. (2003) Proc. Natl. Acad. Sci. USA 100, 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hohmann, H., Hellquist, B. & Van Eldik, R. (1992) Inorg. Chem. 31, 345–351. [Google Scholar]
- 43.Jennerwein, M. & Andrews, P. A. (1995) Drug Metab. Dispos. 23, 178–184. [PubMed] [Google Scholar]
- 44.Novakova, O., Chen, H., Vrana, O., Rodger, A., Sadler, P. J. & Brabec, V. (2003) Biochemistry 42, 11544–11554. [DOI] [PubMed] [Google Scholar]
- 45.Martin, R. B. (1999) in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. Lippert, B. (Wiley–VCH, Zurich), pp. 183–205.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.