

Abstract
Informative genes from microarray data can be used to construct prediction model and investigate biological mechanisms. Differentially expressed genes, the main targets of most gene selection methods, can be classified as single- and multiple-class specific signature genes. Here, we present a novel gene selection algorithm based on a Group Marker Index (GMI), which is intuitive, of low-computational complexity, and efficient in identification of both types of genes. Most gene selection methods identify only single-class specific signature genes and cannot identify multiple-class specific signature genes easily. Our algorithm can detect de novo certain conditions of multiple-class specificity of a gene and makes use of a novel non-parametric indicator to assess the discrimination ability between classes. Our method is effective even when the sample size is small as well as when the class sizes are significantly different. To compare the effectiveness and robustness we formulate an intuitive template-based method and use four well-known datasets. We demonstrate that our algorithm outperforms the template-based method in difficult cases with unbalanced distribution. Moreover, the multiple-class specific genes are good biomarkers and play important roles in biological pathways. Our literature survey supports that the proposed method identifies unique multiple-class specific marker genes (not reported earlier to be related to cancer) in the Central Nervous System data. It also discovers unique biomarkers indicating the intrinsic difference between subtypes of lung cancer. We also associate the pathway information with the multiple-class specific signature genes and cross-reference to published studies. We find that the identified genes participate in the pathways directly involved in cancer development in leukemia data. Our method gives a promising way to find genes that can involve in pathways of multiple diseases and hence opens up the possibility of using an existing drug on other diseases as well as designing a single drug for multiple diseases.
Introduction
Gene selection/biomarker identification methods can be classified as linear and nonlinear methods [1]. A linear method assumes that good biomarkers for a class will be highly expressed (or unexpressed) for that class and unexpressed (or highly expressed) for the rest of the classes [2], [3]. Nonlinear methods identify biomarkers exploiting both linear and nonlinear interactions between classes and genes [4], [5]. Most methods are linear in nature and there are a few non-linear approaches. Nonlinear approaches can discover a small set of discriminatory genes that are good enough for diagnosis of a set of diseases. But the relationship of biomarkers identified by nonlinear methods with different classes may not be easily visualized. Saeys et al. [6] reviewed various methods of biomarker identification and suggested three categories for the existing methodologies. The first one is the filter method category, which ranks genes independently of the classifier that is used. Many linear methods, either univariate or multivariate, e.g., “SAM” [7], “shrinkage t” [8], “correlation-adjusted t” [9] belong to this category. Most of these methods are essentially based on Student's t-test or its modified/adapted forms. In some cases, in conjunction with the t-test, authors have used additional procedures to account for the high-dimensional characteristic of microarray data. In addition to t-test, SAM also uses Wilcoxon's Rank Sum test. In this category, other parametric and non-parametric tests have also been used [10], [11]. The second one is the wrapper method category, which selects genes according to the predictive performance of the associated classifier [12]-[14]. The final category of embedded methods assigns weights to the importance of genes by making use of the internal parameters of the classification model – this is an integrated approach where the feature weighting/selection and classifier design are done simultaneously. The well-known SVM-RFE [4], other SVM-based methods, e.g., MMC-RFE [15], MSVM-RFE [16], SVM-RCE [17], and SVM-RNE [18] all belong to this category. Also, some neural network-based methods, e.g., online FSMLP [5], belong to the same category. The advantages of these wrapper and embedded methods are that they can capture non-linear interactions between genes as well as interaction between genes and the diseases. As a result, such marker genes usually provide a better predictive performance.
Most existing gene selection methods (particularly linear methods) can identify only single-class specific signature genes (i.e., genes that are expressed for one class and unexpressed for the rest of the classes). However, there could be genes which are expressed for a subset of classes (say, for a subgroup of cancers). Such genes are biologically informative genes. Unfortunately, we have failed to locate any study dealing with this important issue. None of the methods discussed earlier can be used easily to identify “multiple-class specific” marker genes. In principle, it may be possible to use methods in the filter category, but it will involve extensive computation. For example, due to the two-class discriminant nature of statistics, such as t-statistic (that is frequently used), further strategies, e.g., one-versus-one (OVO), one-versus-all (OVA), are required to apply these methods in identification of multiple-class specific marker genes. Consequently, to find useful biomarkers, a time-consuming procedure considering all possible combinations of classes has to be performed. Pavlidis and Nobel have proposed an ANOVA and template matching based approach [19] to identify such multiple-class specific biomarkers. But as explained above, to find genes with multiple-class specific signatures, we need to try all possible combinations of classes, which would demand considerable computation. Also, another interesting but time consuming procedure has been proposed in [20] to assign significance to statistically defined expression patterns.
On the other hand, for methods in the embedded and wrapper categories, although we can get better predictive performance, the interactions between the biomarkers and the diseases may not be easy to interpret/understand due to the non-linear nature of interactions. Moreover, to get multiple-class specific biomarkers, we shall still require some post-processing, which may not be easy. Finally, there may be some biomarkers for some specific groups of diseases, but such a method may not recognize/identify those because such methods are driven by minimization of classification error. Hence, there is a need for developing methods/algorithms that can find genes with multiple-class specific signatures.
Here we propose a novel gene selection index, Group Marker Index (GMI), which can identify de novo in a single process both single- and multiple-class specific signature genes (both are called group specific genes). It is computationally efficient in the sense that it does not require computation of the index for all possible subsets of classes. For a K-class data set, GMI is evaluated K-1 times rather than for 2K-2 combinations. GMI is a distribution-free method, which shares the advantage of non-parametric methods and is not influenced by the lack of knowledge about the distribution of data. Furthermore, we use GMI with a repeated random sampling procedure to select candidate marker genes in a more reliable manner. Here a permutation procedure is used to assess the significance of selected marker genes. We have used four multiple-class microarray data sets, which are Small Round Blue Cell Tumors (SRBCT) [21], Leukemia [22], Central Nervous System (CNS) [23], and Lung Cancer [24], to demonstrate the effectiveness of the proposed method. We demonstrate that genes identified by the proposed scheme participate in several important biological processes. Scatter-plots of the identified group specific genes for these data sets also exhibit good discriminating power among classes. Although we could not find any method in the literature addressing this problem, just for the sake of comparison, we have proposed and used an algorithm using a template-based gene selection scheme.
Results
Significance of genes selected by using GMI
In this study, we develop a gene evaluation index named “Group Marker Index (GMI)”, to select biologically significant genes for both two- and multiple-class cancer discrimination problems. Given expression profiles on K different types of cancers (diseases), our GMI-based algorithm tries to discover subsets of the cancers/diseases, if present, that can be discriminated from the remaining ones using gene expressions, as well as, the discriminatory genes. If a gene is highly expressed for a subset containing n classes (n = 1, 2, …, K-1) compared to the remaining (K-n) classes, then we call that gene a level-n discriminatory gene. This set of cancer classes with higher expression values is called the “upper group”. On the other hand, the set of remaining classes with lower expression values is called the “lower group”. The GMI algorithm finds, all level-n (n = 1, 2, …, K-1) genes, if exist. The detailed description of the algorithm is provided in the Materials and Methods section.
The quality of genes selected by using GMI is demonstrated here in two significant ways. First, for each of the four data sets, we present visual assessments of the quality of the top most level-n gene for every level-n selected by GMI. We use a scatter-plot to show the distribution of gene expression values for the top most level-n gene. For the scatter-plot the y-axis expresses the observed gene expression values (normalized in [0,1]) and the x-axis indicates the number and identification of samples in a data set. The samples in different classes are represented by different symbols and colors, which help illustrate the discriminating power of (each) individual GMI-selected gene. For example, each sub-panel in Fig. 1 and Figs. S1, S2, S3 displays a top most level-n gene for different level n in the different data sets. As expected, each such gene appears with high gene expression values in the samples from n classes (i.e., the upper group, infra at Computation of GMI), but with low gene expression values in the samples of the remaining classes/subgroups (i.e., the lower group). Also, every top most level-n gene demonstrates good separation and low overlap between samples belonging to upper and lower groups.
Figure 1. Scatter-plots of the top most gene of each level in the SRBCT data set.

Panels (a), (b) and (c) are the scatter-plots of the top most gene of level-1, level-2, and level-3, respectively. The top most genes are WAS (236282), PTPN12 (774502) and GSTA4 (504791), respectively. There are four classes in the SRBCT data set: Ewing sarcomas (EWS), Burkitt lymphomas (BL), neuroblastomas (NB), and rhabdomyosarcomas (RMS).
In addition, we have further tabulated (Table 1 and Tables S1, S2, S3) the following basic information for the top 10 genes, which are individually selected using GMI from each level of discrimination: probe name, official gene symbol, class-labels in upper group and lower group, frequency of appearance of the gene in the list of top 10 genes, averaged GMI value, p-value and q-value obtained from the permutation test mentioned in the Materials and Methods section. These tables further provide us with several interesting conclusions: (a) We find that all GMI-selected genes have very low p- and q-values (in many cases it is so low that it is represented as zero) suggesting that these genes carry statistically significant multiple-class specific signatures. (b) For a given level of discrimination, the genes with quite high GMI values and appearing with high frequencies are considered better group-specific biomarkers. (c) We use the GMI value to represent a measure of discrimination for a gene between upper and lower groups. Thus, a gene with a higher GMI value implies its higher discriminating power between upper and lower groups. This information can be used for comparison of discriminating power of genes at different levels in the same data set. As demonstrated by the GMI values in Table 1 for the SRBCT data set, the discriminating power of level-2 genes is weaker than that of level-3 genes, and the discriminating power of level-3 genes is weaker than that of level-1 genes. Also as shown by the GMI values set forth in Table S3 for the Lung Cancer data set, the discriminating power of level-3 and level-4 genes is weaker than that of level-1 and level-2 genes. In addition, such phenomena can also be validated by the scatter-plot of the top most gene of each level for all data sets (Fig. 1 and Figs. S1, S2, S3). For example, WAS, the top most level-1 gene in the SRBCT data set, has a GMI value of 2.98 which is higher than the GMI value, 1.02, of PTPN12, the top most gene of level-2 in the same data set. From Fig. 1, we can observe that the discriminating ability of the WAS gene is much stronger than that of the PTPN12 gene because the WAS gene has no overlapped sample between upper and lower groups, whereas the PTPN12 gene has some overlapped samples. Hence, the visual illustration of the top most genes is consistent with the corresponding GMI values. (d) The GMI values can provide an objective assessment of the discriminating power even between data sets. For example, VAMP2, the top most level-1 gene in the Lung Cancer data set, has a GMI value of 7.19, showing discriminating power which appears even stronger than WAS, the top most level-1 gene in the SRBCT data set. Figure 1 and Figure S3 also reveal the stronger discriminating power of VAMP2 over WAS because VAMP2 also has no overlapped samples between upper and lower groups, and gets a bigger numerator for computing GMI (a higher value of the closest separation between a class in the upper group and a class in the lower group; i.e., the difference between the mean values of classes Cn (s) and Cn +1(s), or μSep = μn (s) – μn +1(s) in Eq. (1) mentioned in the Materials and Methods section). Our results demonstrate that GMI is very effective in identifying group-specific marker genes with clear discriminating power and we could obtain a robust list of candidate genes by conducting the repeated random splitting procedure.
Table 1. Summary of top 10 genes of each level selected by GMI in the SRBCT data set.
| Level | Probe ID | Gene Symbol | Upper Group | Lower Group | Freq. | Ave. GMI value | p-value | q-value |
| 1 | 236282 | WAS | 2 | 143 | 100 | 2.98 | 0 | 0 |
| 770394 | FCGRT | 1 | 432 | 99 | 4.98 | 0 | 0 | |
| 241412 | ELF1 | 2 | 143 | 91 | 2.45 | 0 | 0 | |
| 814260 | FVT1 | 1 | 234 | 76 | 3.65 | 0 | 0 | |
| 377461 | CAV1 | 1 | 432 | 57 | 2.37 | 0 | 0 | |
| 784224 | FGFR4 | 4 | 213 | 53 | 2.43 | 0 | 0 | |
| 812105 | MLLT11 | 3 | 412 | 52 | 2.57 | 0 | 0 | |
| 1435862 | CD99 | 1 | 432 | 50 | 2.14 | 0 | 0 | |
| 183337 | HLA-DMA | 2 | 413 | 42 | 1.78 | 0 | 0 | |
| 796258 | SGCA | 4 | 231 | 37 | 2.02 | 0 | 0 | |
| 2 | 774502 | PTPN12 | 34 | 12 | 95 | 1.02 | 0 | 0 |
| 365826 | GAS1 | 14 | 32 | 75 | 0.80 | 0 | 0 | |
| 784593 | RND3 | 34 | 12 | 72 | 0.75 | 0 | 0 | |
| 812965 | MYC | 21 | 43 | 64 | 0.72 | 0 | 0 | |
| 789182 | PCNA | 23 | 41 | 54 | 0.64 | 0 | 0 | |
| 859359 | TP53I3 | 43 | 12 | 44 | 0.62 | 0 | 0 | |
| 714453 | IL4R | 42 | 13 | 38 | 0.57 | 2.17E-06 | 2.17E-06 | |
| 82903 | (EST) | 21 | 43 | 37 | 0.54 | 2.17E-06 | 2.17E-06 | |
| 159455 | PLD3 | 41 | 32 | 34 | 0.53 | 2.17E-06 | 2.17E-06 | |
| 308163 | YAP1 | 41 | 32 | 32 | 0.53 | 2.17E-06 | 2.17E-06 | |
| 3 | 897164 | CTNNA1 | 134 | 2 | 100 | 1.40 | 0 | 0 |
| 504791 | GSTA4 | 314 | 2 | 100 | 1.89 | 0 | 0 | |
| 897788 | PTPRF | 134 | 2 | 100 | 1.40 | 0 | 0 | |
| 295985 | CDK6 | 342 | 1 | 86 | 1.52 | 0 | 0 | |
| 810057 | CSDA | 142 | 3 | 69 | 1.19 | 0 | 0 | |
| 21652 | CTNNA1 | 143 | 2 | 61 | 0.85 | 0 | 0 | |
| 51320 | DBNDD1 | 341 | 2 | 50 | 0.73 | 0 | 0 | |
| 212542 | PBX1 | 143 | 2 | 48 | 0.59 | 0 | 0 | |
| 813742 | XPO6 | 341 | 2 | 38 | 0.80 | 0 | 0 | |
| 741831 | PLTP | 143 | 2 | 28 | 0.54 | 2.17E-06 | 2.17E-06 |
Ewing sarcomas (EWS), Burkitt lymphomas (BL), neuroblastomas (NB), and rhabdomyosarcomas (RMS) are represented as Group 1 to Group 4 in order.
In addition, the conventional biomarkers for cancers are usually the level-1 genes with single tissue specific expression patterns. The protein products of those marker genes are usually receptors or proteins expressed in the cell surface. GMI can also identify the known biomarkers. For example, FGFR4, the level-1 gene highly expressed in rhabdomyosarcomas (RMS) class, is ranked within the top 10 level-1 genes in the SRBCT data set (Table 1). Multiple studies have reported that FGFR4 is highly expressed in RMS [25]-[27] and the mRNA expression level of FGFR4 is correlated with its protein level [25], [26], [28]. Furthermore, it has been shown that mutationally activated FGFR4 acts as an oncogene [29]. Another level-1 gene ranked within the top 10 for the SRBCT data set, CD99, is highly expressed in Ewing sarcomas (EWS) class. It has been reported that CD99 is highly expressed in all EWS and the engagement of CD99 with anti-CD99 monoclonal antibodies would induce massive apoptosis as well as reduce malignant potential of EWS cells [30]. The deletion of CD99 expression in human EWS cell lines would reduce their abilities of tumorigenesis and metastasis [31]. Moreover, the engagement of CD99 improves the efficiency of the conventional chemotherapeutic agents and reduces tumor growth along with a significant delay of metastasis [32]. These are some of the examples to demonstrate that GMI is capable of identifying the known biomarkers as well as the special type of group biomarkers.
Comparison with template-based method
For a fair comparison of our GMI method with the existing methods for similar purpose, we have selected the most widely used template-matching based method and adapted it using the same iterative procedure as followed for GMI. We call this method as the template-based method (TBM). The detailed steps of the template-based method are described in the Materials and Methods section. First, we have used TBM to select group specific genes in the SRBCT and CNS data sets. Although, for the CNS data set, GMI can identify good level-2 discriminatory genes, it cannot identify good level-2 discriminatory genes for the SRBCT data set (please refer to Table 1 and Table S2). So for the SRBCT data set we want to check if there are useful level-2 discriminatory genes and GMI cannot identify those. For the CNS data set, we want to check whether GMI has already found good level-2 and level-3 discriminatory genes. In other words, we want to check if TBM can identify better genes. For each case, we have compared the top 10 genes identified by GMI and TBM.
Comparing the top 10 level-2 genes in the SRBCT data set (Table 2), we find that both GMI and TBM select PTPN12 (774502) as the top most gene. Therefore, there may be no better level-2 gene in the SRBCT data set. To further check whether GMI miss any good level-2 gene, we focus on those genes which are identified by TBM but not by GMI. Between the two gene lists, there are four common genes. With a careful inspection of scatter-plots of the TBM-selected unique genes, we find that for each of such genes there is a substantial overlap between the upper and the lower groups. For a few of the GMI-selected unique genes, a similar situation arises. Thus to make a better objective evaluation of the two lists, for each gene in Table 2, we have performed a leave-one-out cross validation (LOOCV) with the nearest neighbor classifier (NNC), which enables us to assess the discriminating power of each gene for the two groups of diseases. The resultant accuracies are tabulated in the last column of Table 2. On the average, the GMI-selected unique genes are superior to the TBM-selected unique genes. One GMI-selected unique gene, Probe ID = 365826 (ranked the second by GMI) achieves the highest accuracy of 0.9048! On the other hand, Probe ID = 841620, which is ranked the second and selected only by TBM, achieves the least accuracy of 0.6508! This suggests that GMI selected genes are better discriminator; however, as we have mentioned earlier, finding genes suitable for designing classifiers is not the objective of this study.
Table 2. The comparison of top 10 level-2 genes selected by GMI and TBM in the SRBCT data set.
| Probes | GMI Mean Order | GMI Rank | GMI Freq. | TBM Rank | TBM Template | TBM Freq. | LOOCV NNC Acc. |
| 774502 | (34)(12) | 1 | 95 | 1 | (34)(12) | 83 | 0.8413 |
| 365826 | (14)(32) | 2 | 75 | 33 | (14)(23) | 7 | 0.9048 |
| 784593 | (34)(12) | 3 | 72 | 19 | (34)(12) | 16 | 0.7778 |
| 812965 | (21)(43) | 4 | 64 | 4 | (12)(34) | 65 | 0.7143 |
| 789182 | (23)(41) | 5 | 54 | 5 | (23)(14) | 50 | 0.8730 |
| 859359 | (43)(12) | 6 | 44 | 13 | (34)(12) | 25 | 0.8889 |
| 714453 | (42)(13) | 7 | 38 | 8 | (24)(13) | 40 | 0.7460 |
| 82903 | (21)(43) | 8 | 37 | 32 | (12)(34) | 7 | 0.7460 |
| 159455 | (41)(32) | 9 | 34 | 22 | (14)(23) | 12 | 0.8254 |
| 308163 | (41)(32) | 10 | 32 | 24 | (14)(23) | 11 | 0.7302 |
| 841620 | (13)(42) | 12 | 27 | 2 | (13)(24) | 77 | 0.6508 |
| 789204 | (23)(41) | 16 | 21 | 3 | (23)(14) | 71 | 0.7460 |
| 841641 | (13)(42) | 11 | 28 | 6 | (13)(24) | 47 | 0.7778 |
| 809557 | (23)(14) | 18 | 17 | 7 | (23)(14) | 40 | 0.7460 |
| 782811 | (23)(14) | 33 | 6 | 9 | (23)(14) | 36 | 0.6984 |
| 47542 | (23)(41) | 15 | 22 | 10 | (23)(14) | 35 | 0.7460 |
TBM: Template-based method.
Ewing sarcomas (EWS), Burkitt lymphomas (BL), neuroblastomas (NB), and rhabdomyosarcomas (RMS) are represented as Group 1 to Group 4 in order.
Table S4 reveals that for the CNS data set in the lists of top 10 level-2 genes identified by GMI and TBM there are 8 common genes; while for the level-3 genes there are 5 common genes (Table S5). Most of the genes common to both methods are on the top of the TBM gene list. These findings imply that there may not be better level-2 and level-3 discriminatory genes in the CNS data set. Also, the higher agreement between the gene lists identified by GMI and TBM in this data set with more balanced distribution of number of samples over classes may indicate that difference in sample sizes between classes can affect the gene selection.
To investigate the effect of variation of sample sizes between classes, we have compared the gene lists produced by GMI and TBM on Leukemia and Lung Cancer data sets. All classes in the Leukemia data set have comparable sample size. On the other hand, the Lung Cancer data set has one class with a relatively large sample size. We compare the top 10 genes for these two data sets. For the Leukemia data set, considering the level-2 genes we find that seven of the top 10 genes are common in the two lists. Moreover, both lists have the same genes at the top (Table S6). This high percentage of common genes in the two lists might be taken as an indicator that TBM would identify genes similar to those identified by GMI in cases where there is not much variation between sample sizes from different classes.
For the Lung Cancer data set, we consider both level-2 and level-3 discriminatory genes. In the case of level-2 genes, we have three common genes in the top 10 genes identified by GMI and TBM (Table S7). In order to assess the quality of the genes identified by GMI and TBM, we have compared the scatter-plots of those genes, which are identified by only one of the methods. A careful inspection of the scatter-plots suggests that the genes identified only by TBM are not better genes than those identified only by GMI for the purpose of discrimination (see File S1). Furthermore, TAGLN3 (32650_at), which is ranked by GMI as the top most level-2 gene with the highest frequency of selection, is not in the top 10 gene list selected by TBM. But the scatter-plot of TAGLN3 (Figure S3b) reveals that it is definitely a good level-2 gene. Concordant results are also revealed by the single gene LOOCV accuracy as depicted in Table S7, where TAGLN3 achieves the perfect accuracy. While comparing the level-3 discriminatory genes, we have found that there are three common genes between the lists of top 10 genes identified by GMI and TBM (Table S8). Four of the top 10 genes (2nd, 3rd, 5th and 10th) selected by TBM appear much like level-2 genes (see Figs. 2a-d ). The top sixth and eighth genes selected by TBM appear more like level-1 genes (see Figs. 2e–f ). Unlike genes selected by TBM, the scatter-plots of the top genes identified by GMI reveal that these genes are good level-3 discriminatory genes. On the average, the single gene LOOCV accuracies of GMI-selected unique genes are higher than the accuracies of TBM-selected unique genes.
Figure 2. The level-2-like and level-1-like genes ranked within top 10 level-3 genes by template-based method in the Lung Cancer data set.

Panels (a), (b), (c) and (d) are the scatter-plots of the level-2-like genes. Panels (e) and (f) are the scatter-plots of the level-1-like genes.
We find that when the class sizes are balanced, more genes are found common in the top 10 level-n genes produced by GMI and TBM. When the class sizes are widely different, the lists of top 10 level-n genes are significantly different. And GMI is found to identify better discriminatory genes. A natural question arises, why? A possible reason for this may be the fact that correlation value for a gene with the designated template (used in TBM) is quite sensitive to the relative sizes of different classes in the training data. For example, even if the sizes of different classes remain the same, but the number of samples from one class keeps on increasing, the correlation value exhibit a monotonic behavior. This is illustrated in Fig. 3. Suppose in a K-class problem, class 1 has n1 samples, class 2 has n2 samples and the remaining classes together have n3 samples. Suppose for a gene g, all samples from class 1 and 2 are highly expressed (with gene expression value of 1), while for the other classes the gene g is unexpressed (i.e., the gene expression value is zero). In this case, the correlation for the gene g with the ideal vector for class 1 (the ideal vector will have expression of 1 for class 1 and 0 for all other classes) will be 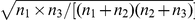 . It can be easily seen that as n3 increases with more and more unexpressed samples keeping the nature of the n1 and n2 samples from class 1 and 2 unaltered, the correlation goes to
. It can be easily seen that as n3 increases with more and more unexpressed samples keeping the nature of the n1 and n2 samples from class 1 and 2 unaltered, the correlation goes to 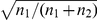 . The filled circle in Fig. 3 illustrates this when n1 = 60, n2 = 15 and n3 varies from 20 to 300. Note that, when n3 = 20, the correlation value is 0.68 but that increases to 0.85 when n3 becomes 140, although neither the expression profiles for class 1 and class 2 change nor the expression profile of the remaining classes changes, and for a multiple-class data 140 is not a big number. Thus, the template-based method, which selects genes based on correlation value, sometimes is not sensitive enough to reflect the desired behavior of samples. This will lead to improper ranking.
. The filled circle in Fig. 3 illustrates this when n1 = 60, n2 = 15 and n3 varies from 20 to 300. Note that, when n3 = 20, the correlation value is 0.68 but that increases to 0.85 when n3 becomes 140, although neither the expression profiles for class 1 and class 2 change nor the expression profile of the remaining classes changes, and for a multiple-class data 140 is not a big number. Thus, the template-based method, which selects genes based on correlation value, sometimes is not sensitive enough to reflect the desired behavior of samples. This will lead to improper ranking.
Figure 3. Effect of sample size on Pearson's correlation coefficient values.

Following the same experimental design, we add one extra class (class (K+1)) with only five samples, which is either up-regulated or down-regulated. The template is so set that samples in class 1 and class (K+1) are highly expressed while unexpressed in the remaining classes. When class (K+1) is up-regulated, the tendency of the resultant correlation values (shown by the symbol “+” in Fig. 3) is almost the same as that of the K-class case. In case, class (K+1) is down-regulated, the nature of the resultant correlation values (shown by triangles in Fig. 3) is still similar to the previous two cases with correlation values in a slightly lower region. In these two (K+1)-class cases, most samples are unexpressed, which are contributed by the pool (class 3 to class K) with n3 samples. Therefore, it has practically no effect when we set class (K+1) up-regulated. On the other hand, there is a little influence when class (K+1) is down-regulated. However, as the number of samples increases, the impact becomes less and less.
The level 2-like genes selected by TBM in the Lung Cancer data set (Figs. 2a–d) are good examples to show that this effect indeed happens in real data. All of those four genes are selected based on the template with high expression for classes 2, 3, and 5 (please refer to Table S8). However, the Lung Cancer data set is quite unbalanced in the number of samples between different classes. There are only six samples for class 3, which contributes a little in computing the correlation value of TBM (only four samples are randomly selected during the repeated random splitting procedure). Therefore, irrespective of whether class 3 is highly expressed or unexpressed, the contribution of class 3 in correlation may not be significant (Figs. 2b–d ). Even for class 2 (Normal) which has 17 samples, the selection by TBM suffers from the same problem (Fig. 2a). Similarly, the level 1-like genes (Figs. 2e–f ) can also support this effect.
Biological Relevance of some level-2 and level-3 biomarkers
CNS data set
The first 4 genes in level-2 are highly expressed in human cerebellar tumors and malignant glioma (i.e., Ncer and MGlio; upper group representing 4 and 2) and are practically unexpressed in the rest of the classes (lower group consisting of 1, 3 and 5). Interestingly, six of the top genes in level-2 (PRUNE2, TIMP4, TMOD1, ADORA1, NEUROD1 and C1orf61/Croc4) with GMI score ranging from 2.38 to 1.17 are primarily, if not solely, involved in cytoskeleton maintenance. Perturbation in the expression levels of these genes is likely to affect morphological, structural and functional integrity of the cell. For example, PRUNE2 gene is over-expressed in prostate cancer and down-regulates Rho-A and Rho-C that are involved in actin polymerization and oncogenic transformation [33], [34]. TIMP4 inhibits tumor progression by inhibiting cell matrix degradation by endopeptidase MMP-2 [35]. TMOD1 regulates actin filament dynamics [36]. On the other hand, C1orf61/Croc4 positively controls c-fos activity and the later one up-regulates actin expression [37], [38]. Moreover, NEUROD1, a transcriptional factor, controls transcription of cytoskeletal genes [39]. In cancer, one of the frequently affected pathways is the cytoskeletal structural organization and the genes in this pathway are dys-regulated but not necessarily mutated. For example, cofilin expression is frequently increased in glioblastoma and ovarian cancer [40], [41] whereas cortactin is often over-expressed in breast cancer and squamous carcinoma of head and neck [42], [43]. Literature search revealed that none of the level-2 genes are ever mutated in any cancer but rather work to protect the cytoskeletal integrity of the cell. Over-expression of these genes in human cerebellar tumors and malignant glioma may reflect as an innate attempt of the cell to counteract the process of tumor transformation.
Earlier we proposed GDI system (Gene Dominant/Dormant Index) that selects two specific types of gene, i.e., dominant and dormant genes wherein the former is up-regulated in one specific class and down-regulated in the remaining classes, and the latter is down-regulated in one specific class but up-regulated in the rest of the classes [1]. Although different cancers, according to their tissue of origin, do differ in their mode of action through regulation and/or dys-regulation of various physiological pathways, they do also share some common genes/pathways in the formation of cancers. In that sense, GMI-discovered genes that are up-regulated in more than one-class of cancer (here we can also termed them as co-dominant) may provide additional biological insights of how various cancers might be related and probable molecular pathways involved in them.
In case of the level-3 genes, it appears that they are functionally diverse. However, the first two genes share a common cytoskeletal pathway. For example, FEZ1 (GMI score 3.95), highly expressed in the group {4, 2, 5}, is a brain specific cytoskeletal regulatory protein associated with microtubule and in various tumors it is either deleted and/or mutated [44]. But here in malignant glioma, human cerebellar tumor and primitive neuroectodermal tumor (PNET) the expression of FEZ1 is increased signifying a failed attempt of the cell to contain the tumor growth. Recent study shows that FRG1 (GMI value 2.07, up-regulated in group {1, 4, 3}) a multifunctional protein, specially binds to F-actin and its misregulation leads to facioscapulohumeral muscular dystrophy (FSHD) [45]. HMGN2 with GMI value of 1.11 (up-regulated in group {1, 3, 5}) is a highly conserved nucleosomal protein involved in unfolding higher-order chromatin structure and acts as an impediment for cell migration [46], [47]. VAT1 with GMI value of 1.14 (up-regulated in group {3, 1, 2}) controls the storage and release of neurotransmitters in the nerve terminal [48]. Importantly, none of these top-5 genes in both level-2 (except PRUNE2) and level-3 are ever reported as biomarkers (over-expressed and/or under-expressed) for any type of cancer. In that sense, our analysis demonstrates that these co-dominant genes may be used as unique signature genes in defining the respective tumor groups.
Lung Cancer data set
Level-2 and level-3 discriminatory genes in the Lung Cancer data set are quite interesting. As in the case for CNS data set, the four top level-2 discriminatory genes with GMI values ranging from 3.45 to 1.58 are primarily involved in regulation of cytoskeleton that controls vesicular trafficking in golgi bodies [49]. These genes are basically co-dominant in classes 5 and 3, representing pulmonary carcinoids (COID) and small cell lung cancer (SCLC), respectively. TAGLN3 associates with and regulates F-actin, α-tubulin, tau and MAP2 [50]. CRMP1 is a phosphoprotein and controls microtubules [51]. NCAM1 is a classical cell adhesion molecule that interacts with a number of cytoskeletal proteins and regulates cell architecture. INSM1 gene encodes a zinc finger DNA-binding domain and a putative prohormone domain. This gene is a sensitive marker for neuroendocrine differentiation of human lung tumors [52]. Interestingly, INSM1 acts as a transcriptional repressor of NEUROD that is involved in regulating cytoskeleton genes [53]. It is important to note that four of the first five genes in level-2 (TGLN3, CRMP1, NCAM1 and SCAMP5) are supposed to be neuron-specific [54]-[57]. But our analysis shows that these neuron-specific genes are aberrantly expressed in COID and SCLC class of lung tumor, most likely as a result of mis-functioning of SWI/SNF complex [58]. The fact that the top 4 level-2 discriminatory genes both in CNS and Lung Cancer data sets are involved in the same molecular pathway might be a pure coincidence or else it could be that pathways of cancer, irrespective of types, are frequently involved dys-regulating genes associated with cytoskeleton architecture.
On the other hand, the top 6 level-3 discriminatory genes are basically involved in calcium signaling; however, they bear quite a low GMI values ranging from 1.25 to 0.34. These genes, except SEC14L1, are primarily co-dominant in the classes 4, 1 and 2, i.e., squamous cell carcinomas (SQ), lung adenocarcinomas (Adeno) and normal lung (Normal). S100A11 and S100A10 are calcium binding proteins and their increased expression are often observed in colorectal and prostate cancer, including non-small cell lung cancer [59], [60]. VAMP8 is involved in calcium-dependent exocytosis process and SEC14L1 is a membrane trafficking protein, and none of them is linked to cancer pathways. On the other hand, LGALS3, an IgE-lectin binding protein, is often abundantly expressed in thyroid cancer, hepatocellular carcinoma and in non-small cell lung cancer [61], [62]. FAM38 is involved in intracellular calcium release [63]. Calcium signaling plays very important role in cancer in mediating angiogenesis steps such as invasion, adhesion and tumor cell migration. In many cancers, calcium signaling genes are up-regulated. The fact that the aforementioned genes are also up-regulated in normal lung (class 2) indicates that intact calcium signaling pathway may be necessary for tumorigenesis process in these cancer subgroups.
In a nutshell GMI method establishes that cytoskeleton regulating genes are aberrantly expressed in pulmonary carcinoids (COID) and small cell lung cancer (SCLC) whereas calcium signaling pathway genes are active in the rest of the classes, i.e., squamous cell carcinomas (SQ), adenocarcinomas (Adeno), and normal lung (Normal).
Typical microarray data analysis finds markers that can differentiate one class of cancer from the others, while GMI can find markers, which exhibit similar expression pattern in a group of cancers and can distinguish one group of cancers from another group of cancers. Thus, the importance of our GMI algorithm is in identifying de novo group specific genes in an automatic manner. When we have samples from multiple cancers/diseases with less knowledge about the relationship between them, GMI could be very useful to discover a group (subset) of cancers, if exists, that can be discriminated from the remaining set of cancers using expression pattern of one or more genes. This in turn can help us to understand the relationship between subsets of cancers/diseases groups via functional analysis of the GMI-identified group specific genes. Further, existence and discovery of such group markers open up the possibility of finding common drug targets for different diseases as well as possibility of using drugs designed for one disease to cure another disease.
KEGG pathway analysis
After analyzing the relationship between a single gene and multiple diseases, we attempt to explore the relationship between the groups of discriminatory genes and diseases. For this, instead of considering level-1 discriminatory genes, which may be identified by all gene selection methods, we focus on the level-2 discriminatory genes first. We try to identify related biological pathways in which the level-2 discriminatory genes are involved by cross-referencing to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [64]-[66].
We take the advantage of the abundant number of genes in the Leukemia and Lung Cancer data sets in the data selection for pathway analysis. For the Leukemia data set, we consider all level-2 genes with p-values smaller or equal to 0.0001. Subsequently, we divide these level-2 genes into three different gene lists based on the composition of the upper group. The numbers of genes, actually probe sets, in each list were: 712 for the upper group composed of acute lymphoblastic leukemia (ALL) and mixed-lineage Leukemia (MLL), which we call ALL-MLL list; 3 for the upper group composed of ALL and acute myelogenous leukemia (AML), which is termed as ALL-AML list; 100 for the upper group composed of MLL and AML (MLL-AML list). Next, we use the functional annotation tool provided by the database for annotation, visualization and integrated discovery (DAVID ver. 6.7) [67], [68] to cross-reference to the KEGG database (accepted default parameters).
For the ALL-MLL list, 709 probe sets that belong to Homo sapiens, are selected and the default background is used for the analysis. A total of 665 DAVID IDs are converted and 228 probe sets are involved in the KEGG pathway category. The output results provided by DAVID are summarized in Table 3. The identified pathways/annotation terms are tabulated in the first column (Term). The total number of genes (Count) and the corresponding percentage (%) in our gene list involved in each pathway along with the modified Fisher Exact p-value (p-value), and other enrichment quantitative measurements (Fold Enrichment, Bonferroni, Benjamini, FDR) are also included in Table 3. As shown in Table 3, there are four pathways with EASE Score, the modified Fisher Exact p-value, smaller than 0.01. These are spliceosome, B-cell receptor signaling, basal transcription factors and inositol phosphate metabolism pathways. Literature search revealed that these pathways are often impaired in ALL and MLL development [69]–[73]. On the other hand, if we consider EASE Score smaller than 0.05, then 15 pathways are identified. The full table with probe set lists is provided in Table S9. Most of these additional pathways are involved in cancer as evident by their name such as colorectal cancer pathways, base excision repair, mismatch repair, nucleotide excision repair, and more specifically involvement of chronic myeloid leukemia pathway is noteworthy. Interestingly, pathways like fatty acid metabolism, phosphatidylionositol signaling and ubiquitin mediated proteolysis that at first sight seemed to be remotely associated with ALL or MLL but are indeed found to be very much molecularly involved in the pathogenesis of ALL and MLL as revealed by literature search [74]–[77].
Table 3. Summarization of the identified pathways related to the level-2 discriminatory genes in the Leukemia data set.
| Term | Count | % | p-value | Fold Enrichment | Bonferroni | Benjamini | FDR |
| ALL-MLL List | |||||||
| hsa03040:Spliceosome | 21 | 3.1579 | 5.38E-07 | 3.7171 | 8.23E-05 | 8.23E-05 | 6.44E-04 |
| hsa04662:B cell receptor signaling pathway | 14 | 2.1053 | 2.13E-05 | 4.1632 | 0.0033 | 0.0016 | 0.0256 |
| hsa03022:Basal transcription factors | 7 | 1.0526 | 0.0040 | 4.4605 | 0.4623 | 0.1868 | 4.7433 |
| hsa00562:Inositol phosphate metabolism | 8 | 1.2030 | 0.0095 | 3.3041 | 0.7685 | 0.3064 | 10.8272 |
| hsa05210:Colorectal cancer | 10 | 1.5038 | 0.0120 | 2.6551 | 0.8413 | 0.3080 | 13.4271 |
| hsa03420:Nucleotide excision repair | 7 | 1.0526 | 0.0126 | 3.5481 | 0.8565 | 0.2765 | 14.1065 |
| hsa04910:Insulin signaling pathway | 13 | 1.9549 | 0.0168 | 2.1477 | 0.9248 | 0.3091 | 18.3459 |
| hsa03430:Mismatch repair | 5 | 0.7519 | 0.0175 | 4.8484 | 0.9333 | 0.2871 | 19.1067 |
| hsa05220:Chronic myeloid leukemia | 9 | 1.3534 | 0.0178 | 2.6763 | 0.9363 | 0.2636 | 19.3979 |
| hsa03410:Base excision repair | 6 | 0.9023 | 0.0184 | 3.8233 | 0.9419 | 0.2477 | 19.9781 |
| hsa05340:Primary immunodeficiency | 6 | 0.9023 | 0.0184 | 3.8233 | 0.9419 | 0.2477 | 19.9781 |
| hsa04660:T cell receptor signaling pathway | 11 | 1.6541 | 0.0216 | 2.2716 | 0.9646 | 0.2620 | 23.0243 |
| hsa00071:Fatty acid metabolism | 6 | 0.9023 | 0.0312 | 3.3454 | 0.9922 | 0.3327 | 31.6244 |
| hsa04120:Ubiquitin mediated proteolysis | 12 | 1.8045 | 0.0414 | 1.9535 | 0.9985 | 0.3923 | 39.7743 |
| hsa04070:Phosphatidylinositol signaling system | 8 | 1.2030 | 0.0459 | 2.4111 | 0.9992 | 0.4014 | 43.0280 |
| MLL-AML List | |||||||
| hsa05221:Acute myeloid leukemia | 4 | 4.4944 | 0.0055 | 10.6270 | 0.3252 | 0.3252 | 5.5876 |
| hsa04662:B cell receptor signaling pathway | 4 | 4.4944 | 0.0112 | 8.2182 | 0.5520 | 0.3307 | 11.0767 |
| hsa04664:Fc epsilon RI signaling pathway | 4 | 4.4944 | 0.0125 | 7.9021 | 0.5909 | 0.2577 | 12.2495 |
| hsa04062:Chemokine signaling pathway | 5 | 5.6180 | 0.0285 | 4.1201 | 0.8715 | 0.4013 | 25.9129 |
The contents of Tables 3- 5 are the output results provided by DAVID. The first column (Term) contains the identified KEGG pathways. The second column (Count) indicates the total number of genes in our gene list which is involved in each pathway. The third column (%) shows the same information as shown by the second column but using percentage. The following columns (p-value, Fold Enrichment, Bonferroni, Benjamini, FDR) represent the modified Fisher Exact p-value and other enrichment quantitative measurements, respectively. For more detailed information, please refer to DAVID[67], [68].
Using the same procedure, 98 probe sets belonging to Homo sapiens and one probe set belonging to Mus musculus are selected for the MLL-AML list. These probe sets are mapped to 89 DAVID IDs and only 33 probe sets are involved in the KEGG pathway category. Befitting our method of analysis, the only pathway identified by the functional annotation tool with EASE Score smaller than 0.01 is the Acute Myeloid Leukemia pathway (KEGG entry ID: hsa05221). This attests that our method of analysis may be relevant in identifying the gene(s) signatures for distinguishing subtypes of cancer. Additional three pathways, B cell signaling, Chemokine signaling and Fc epsilom R1 signaling, which have passed the EASE Score criterion (p-value < 0.05) are also shown in the Table 3. These are all well documented pathways that are impaired in AML and/or MLL cancer [78]-[80]. Moreover, MLL is a special ALL group, which carries MLL gene translocation. The authors of the Leukemia data set [22] collected ALL and MLL samples from the individuals diagnosed as CD19+ B-precursor ALL without and with MLL translocation, respectively. Those well-known B cell marker genes, e.g., CD19, CD81, CD79A, CD79B, are all contained in the ALL-MLL list. Some other genes, which have been discussed by the authors [22] e.g., IL7R, DNTT, TCF3, POU2AF1 and SMARCA4, are also listed in the ALL-MLL list. The rest of genes in the ALL-MLL list are related to B cell proliferation, phosphorylation, DNA replication, tumor development. On the other hand, the myeloid-specific genes e.g., CCNA1, SERPINB1, RNASE3, and some other genes that are discussed by the authors who published the data set e.g., CD44, HOXA9, HOXA5, SPN, LGALS1, ANXA1, ANXA2, are contained in the MLL-AML list. The only three genes exhibiting common up-regulated pattern in ALL and AML are RYK, SCHIP1, and YESP1. The long list of level-2 discriminatory genes for the ALL and MLL group suggests that between the three classes, the ALL and MLL group share more similar physiological properties. MLL represents mixed-lineage features yet we can find some similarities between MLL and AML. However, ALL and AML are distinguished from each other. In a nutshell, many of the level-2 discriminatory genes identified in our analysis for leukemia class-specific signatures are directly involved in pathways that lead to the development of leukemia. We believe that GMI would be helpful in understanding the relationship between unknown classes.
Furthermore, we find that the B cell receptor signaling pathway was identified by both ALL-MLL list and MLL-AML list from Table 3. To further investigate this pathway, we downloaded the figure of this pathway from KEGG [64]–[66] and labeled the level-2 genes using the same principle as used by DAVID [67], [68]. The modified figure is shown in Fig. S4. In Figure S4, those genes, which are relatively up-regulated in the ALL-MLL group, are labeled with red stars. On the other hand, those genes, which are up-regulated in MLL-AML group, are labeled with blue stars. We may interpret those red star genes as lymphoblastic genes because of the relatively up-regulated expression in both ALL and MLL but down-regulated expression in AML. Subsequently, those blue star genes can be treated as myelogenous genes because of their relatively lower expression in ALL. As summarized by KEGG [64]–[66], the activation of this pathway will involve in B cell proliferation, differentiation and Ig production as well as other processes. Thus, we find that many genes involved in this pathway are labeled with red stars (highly expressed in the CD19+ B-precursor ALL and MLL). We may interpret that the activation of CD79A (Igα) and CD79B (Igβ) with the co-simulators CD81 and CD19 triggers the activation of B cell signaling pathway in ALL and MLL. Furthermore, the co-inhibitor LIRB3 (PIR-B) is also relatively down-regulated in ALL which could be treated as a positive factor for activating the B cell signaling pathway. Notably, the relatively lower expression of Rac in ALL may play an important role of lymphoblastic leukemia. In murine study, it has been demonstrated that Rac genes are important for appropriate positioning of hematopoietic stem cells (HSCs) within the bone marrow microenvironment. The deletion of both Rac1 and Rac2 murine alleles would lead to a massive egress of HSCs into the blood from the marrow [81], [82]. Thus, we may understand the role of B cell signaling pathway between those different groups of leukemia. This is an example to demonstrate how multiple level-2 (in general level-k) genes can be useful to interpret the observations and to understand the mechanisms behind. We believe that GMI algorithm will be a useful algorithm to the community.
In the Lung Cancer data set, we select all probe sets with p-value smaller than or equal to 0.0001 and group these probe sets into different gene lists for different compositions of the upper group. In the results shown in Table 4, there are five combinations in which we can identify related pathways (EASE Score < 0.05). These five combinations are Adeno-Normal, Adeno-SQ, Normal-SCLC, SCLC-SQ and Normal-COID. Each of these combinations belongs to the upper group. These pathways associated with SCLC-COID list are independently tabulated in Table 5. The full tables with probe set lists are also provided in Table S10 and Table S11. Out of these five combinations two combinations, Adeno-SQ and SCLC-SQ are important in order to molecularly distinguish four classes of lung cancer. In SCLC-COID group out of 27 pathways identified, 10 pathways are directly involved in cancer as their name suggests and additional pathways like splicesome, phosphatidylinositol signaling, calcium signaling and cell cycle pathways are also participatory in cancer process as discussed earlier. Importantly, the pathways like wnt, ErbB, MAPK, autophagy and Jak-Stat signaling are all well established in cancer development process. However, it is presently unclear as why Alzheimer's, Type II Diabetes, Long-term potentiation and Neurotrophin signaling pathways genes are up-regulated in SCLC-COID groups since their roles in cancer are not well founded. In the Adeno-SQ list, genes belonging to pathways of Ribosome, ECM receptor interaction and Focal adhesion were found to be up-regulated. Literature search revealed that all of these pathways are more or less compromised in Adeno-SQ [83]-[86].
Table 4. Summarization of the identified pathways related to the level-2 discriminatory genes in the Lung Cancer data set (Part I).
| Term | Count | % | p-value | Fold Enrichment | Bonferroni | Benjamini | FDR |
| Adeno-Normal List | |||||||
| hsa05416:Viral myocarditis | 10 | 9.0909 | 1.17E-07 | 11.5516 | 1.17E-05 | 1.17E-05 | 1.30E-04 |
| hsa05330:Allograft rejection | 8 | 7.2727 | 1.60E-07 | 18.2258 | 1.60E-05 | 7.99E-06 | 1.77E-04 |
| hsa05332:Graft-versus-host disease | 8 | 7.2727 | 2.86E-07 | 16.8238 | 2.86E-05 | 9.55E-06 | 3.18E-04 |
| hsa04940:Type I diabetes mellitus | 8 | 7.2727 | 4.88E-07 | 15.6221 | 4.88E-05 | 1.22E-05 | 5.42E-04 |
| hsa05320:Autoimmune thyroid disease | 8 | 7.2727 | 1.93E-06 | 12.8653 | 1.93E-04 | 3.85E-05 | 0.0021 |
| hsa04612:Antigen processing and presentation | 9 | 8.1818 | 5.23E-06 | 8.8933 | 5.23E-04 | 8.71E-05 | 0.0058 |
| hsa05310:Asthma | 6 | 5.4545 | 2.00E-05 | 16.9689 | 0.0020 | 2.86E-04 | 0.0222 |
| hsa04514:Cell adhesion molecules (CAMs) | 9 | 8.1818 | 1.54E-04 | 5.5920 | 0.0153 | 0.0019 | 0.1711 |
| hsa05322:Systemic lupus erythematosus | 8 | 7.2727 | 1.58E-04 | 6.6276 | 0.0157 | 0.0018 | 0.1754 |
| hsa04672:Intestinal immune network for IgA production | 6 | 5.4545 | 2.68E-04 | 10.0428 | 0.0264 | 0.0027 | 0.2966 |
| hsa00590:Arachidonic acid metabolism | 6 | 5.4545 | 5.03E-04 | 8.7874 | 0.0490 | 0.0046 | 0.5564 |
| hsa04640:Hematopoietic cell lineage | 7 | 6.3636 | 5.18E-04 | 6.6757 | 0.0505 | 0.0043 | 0.5731 |
| hsa04142:Lysosome | 7 | 6.3636 | 0.0026 | 4.9069 | 0.2279 | 0.0197 | 2.8294 |
| hsa04210:Apoptosis | 5 | 4.5455 | 0.0199 | 4.7136 | 0.8660 | 0.1338 | 19.9982 |
| hsa04610:Complement and coagulation cascades | 4 | 3.6364 | 0.0492 | 4.7546 | 0.9936 | 0.2858 | 42.8973 |
| Adeno-SQ List | |||||||
| hsa03010:Ribosome | 7 | 14.2857 | 4.68E-06 | 14.6121 | 0.0002 | 0.0002 | 0.0042 |
| hsa04512:ECM-receptor interaction | 6 | 12.2449 | 6.62E-05 | 12.9719 | 0.0025 | 0.0013 | 0.0598 |
| hsa04510:Focal adhesion | 4 | 8.1633 | 0.0888 | 3.6141 | 0.9708 | 0.6922 | 56.8582 |
| Normal-SCLC List | |||||||
| hsa04510:Focal adhesion | 3 | 10.7143 | 0.0286 | 9.4869 | 0.4567 | 0.4567 | 20.0947 |
| Normal-COID List | |||||||
| hsa04142:Lysosome | 5 | 3.9683 | 3.09E-02 | 4.1001 | 9.26E-01 | 9.26E-01 | 2.85E+01 |
| SCLC-SQ List | |||||||
| hsa03040:Spliceosome | 12 | 12.5000 | 1.83E-08 | 9.6857 | 9.15E-07 | 9.15E-07 | 1.76E-05 |
| hsa03030:DNA replication | 8 | 8.3333 | 3.34E-08 | 22.6000 | 1.67E-06 | 8.36E-07 | 3.22E-05 |
| hsa04110:Cell cycle | 10 | 10.4167 | 2.20E-06 | 8.1360 | 1.10E-04 | 3.67E-05 | 0.0021 |
| hsa03410:Base excision repair | 6 | 6.2500 | 1.76E-05 | 17.4343 | 8.81E-04 | 2.20E-04 | 0.0170 |
| hsa00670:One carbon pool by folate | 3 | 3.1250 | 0.0100 | 19.0687 | 0.3955 | 0.0958 | 9.2400 |
Table 5. Summarization of the identified pathways related to the level-2 discriminatory genes in the Lung Cancer data set (Part II).
| Term | Count | % | p-value | Fold Enrichment | Bonferroni | Benjamini | FDR |
| SCLC-COID List | |||||||
| hsa04020:Calcium signaling pathway | 55 | 2.0992 | 6.44E-06 | 1.8058 | 1.23E-03 | 1.23E-03 | 0.0080 |
| hsa05223:Non-small cell lung cancer | 24 | 0.9160 | 1.06E-05 | 2.5682 | 0.0020 | 1.01E-03 | 0.0132 |
| hsa03040:Spliceosome | 40 | 1.5267 | 1.01E-04 | 1.8344 | 0.0191 | 0.0064 | 0.1256 |
| hsa04070:Phosphatidylinositol signaling system | 27 | 1.0305 | 1.54E-04 | 2.1083 | 0.0289 | 0.0073 | 0.1909 |
| hsa04722:Neurotrophin signaling pathway | 39 | 1.4885 | 1.56E-04 | 1.8174 | 0.0294 | 0.0059 | 0.1941 |
| hsa04210:Apoptosis | 30 | 1.1450 | 1.89E-04 | 1.9926 | 0.0355 | 0.0060 | 0.2350 |
| hsa05214:Glioma | 24 | 0.9160 | 1.90E-04 | 2.2013 | 0.0357 | 0.0052 | 0.2363 |
| hsa04010:MAPK signaling pathway | 70 | 2.6718 | 1.95E-04 | 1.5149 | 0.0366 | 0.0047 | 0.2428 |
| hsa05218:Melanoma | 26 | 0.9924 | 1.97E-04 | 2.1160 | 0.0369 | 0.0042 | 0.2448 |
| hsa05220:Chronic myeloid leukemia | 27 | 1.0305 | 1.97E-04 | 2.0802 | 0.0370 | 0.0038 | 0.2452 |
| hsa04110:Cell cycle | 38 | 1.4504 | 4.09E-04 | 1.7566 | 0.0752 | 0.0071 | 0.5081 |
| hsa04012:ErbB signaling pathway | 29 | 1.1069 | 4.68E-04 | 1.9261 | 0.0855 | 0.0074 | 0.5806 |
| hsa04930:Type II diabetes mellitus | 19 | 0.7252 | 4.79E-04 | 2.3360 | 0.0874 | 0.0070 | 0.5937 |
| hsa05210:Colorectal cancer | 28 | 1.0687 | 6.01E-04 | 1.9261 | 0.1085 | 0.0082 | 0.7453 |
| hsa05213:Endometrial cancer | 20 | 0.7634 | 6.70E-04 | 2.2225 | 0.1201 | 0.0085 | 0.8297 |
| hsa05200:Pathways in cancer | 80 | 3.0534 | 7.56E-04 | 1.4094 | 0.1345 | 0.0090 | 0.9364 |
| hsa05010:Alzheimer's disease | 45 | 1.7176 | 0.0011 | 1.5953 | 0.1875 | 0.0121 | 1.3434 |
| hsa05215:Prostate cancer | 28 | 1.0687 | 0.0016 | 1.8179 | 0.2646 | 0.0169 | 1.9818 |
| hsa04720:Long-term potentiation | 23 | 0.8779 | 0.0017 | 1.9545 | 0.2790 | 0.0171 | 2.1081 |
| hsa04914:Progesterone-mediated oocyte maturation | 27 | 1.0305 | 0.0021 | 1.8142 | 0.3252 | 0.0195 | 2.5289 |
| hsa04310:Wnt signaling pathway | 41 | 1.5649 | 0.0026 | 1.5690 | 0.3949 | 0.0236 | 3.2188 |
| hsa04114:Oocyte meiosis | 32 | 1.2214 | 0.0028 | 1.6810 | 0.4135 | 0.0240 | 3.4154 |
| hsa05222:Small cell lung cancer | 26 | 0.9924 | 0.0031 | 1.7886 | 0.4524 | 0.0258 | 3.8464 |
| hsa04622:RIG-I-like receptor signaling pathway | 23 | 0.8779 | 0.0032 | 1.8719 | 0.4533 | 0.0248 | 3.8563 |
| hsa05212:Pancreatic cancer | 23 | 0.8779 | 0.0038 | 1.8459 | 0.5188 | 0.0288 | 4.6529 |
| hsa04140:Regulation of autophagy | 14 | 0.5344 | 0.0039 | 2.3114 | 0.5298 | 0.0286 | 4.7956 |
| hsa04630:Jak-STAT signaling pathway | 41 | 1.5649 | 0.0043 | 1.5285 | 0.5645 | 0.0303 | 5.2701 |
To summarize, KEGG pathway analysis establishes that there is as such no specific pathway(s) that can exclusively determine the subgroups of cancer. A plethora of pathways, shared and/or non-shared, is activated in various cancer subgroups. The level-n discriminatory genes found by GMI are important genes that play a major role in multiple cancer pathways.
Discussion
In this study, we emphasize the important role played by multiple-class specific marker genes. We found that most available gene selection algorithms focus on or tend to identify single-class specific signature genes as marker genes. But, multiple-class specific markers may play important roles in biology besides construction of computational prediction systems. The lack of intuitive, easy-to-use methodologies for biologists inspired us to propose a novel gene selection algorithm based on an index called, Group Marker Index (GMI), which is efficient in the identification of both single- and multiple-class specific signature genes from microarray data. For the sake of comparison, a template-based method (TBM) is also formulated and used along with GMI. GMI differs from template-based methods in two aspects. First, to evaluate each gene, GMI uses the ordering of mean gene expression values for all classes that enables it to identify de novo certain combination of classes for each level of discrimination. It does not need to check all combinations of classes and hence it reduces drastically the computation overhead. We have demonstrated that GMI can identify robust and statistically significant marker genes for each level of discrimination using a repeated random-splitting procedure (Table 1 and Tables S1, S2, S3). Second, no prior knowledge is required for template assignment. It is possible for GMI to infer novel relationships between the studied classes/diseases. We have discussed the relevance/biological roles played by several level 2/3 genes. In nutshell, we observed selective dys-regulation of cytoskeleton regulating gene-network primarily at level-2 in both CNS and lung cancer. Secondly, we found that neuron-specific cytoskeleton genes are aberrantly expressed in COID and SCLC tumor of lung. Third, calcium signaling pathway genes are upregulated at level-3 of lung tumor. This method hitherto uncovered the importance of cytoskeleton genes and their use as class-specific markers for cancer diagnosis. In addition, we have also mapped a group of level 2/3 genes to the KEGG pathways. Both results exhibited concordant findings and implied potentially common properties between different classes or cancers. It suggests the possibility of identifying common drug targets between different diseases. It also opens up the possibility of using a specific remedy designed for one disease to cure another disease.
Second, GMI uses a non-parametric indicator, between-class-transition (BCT), to evaluate the discrimination between classes rather than evaluating the similarity between a template and gene expression values. An advantage of exploiting discrimination ability rather than similarity is that it ensures that the selected genes will be able to discriminate between subsets of classes at least to a reasonable extent. Inspired by the work in [19], [20] where authors used Analysis of Variance (ANOVA) along with the template-matching step for their purposes, here we have performed the ANOVA test between gene expression values and class labels for each gene. We find that a gene with a low ANOVA p-value often does not exhibit good discrimination ability between classes. Our experience shows that most genes in a multiple-class microarray data set are evaluated with small p-values by ANOVA test (See Fig. S5 ). Thus, for such a problem the main criterion used for gene selection is essentially the correlation value. Moreover, since TBM relies on correlation, it suffers from an additional problem (as already explained) while applying on the microarray data with unbalanced sample sizes over different classes. We have designed an example with unbalanced samples (Fig. 3) to demonstrate that the class with large sample size biases the resultant correlation value and the class with small sample size may not contribute much to the correlation. This is an inherent limitation of template based methods.
Materials and Methods
Data sets
SRBCT data set
It is a cDNA microarray data set with 63 samples from 4 classes of childhood small round blue cell tumors (SRBCT): 23 Ewing sarcomas (EWS), 8 Burkitt lymphomas (BL), 12 neuroblastomas (NB), and 20 rhabdomyosarcomas (RMS). Each sample is represented by 2308 genes. This data set is available at http://research.nhgri.nih.gov/microarray/Supplement/.
Leukemia data set
This Affymetrix high-density oligonucleotide array data set has 57 samples from 3 classes of leukemia: 20 acute lymphoblastic leukemia (ALL), 17 mixed-lineage leukemia (MLL), 20 acute myelogenous leukemia (AML), each with 12582 genes. This data set is available at http://www.broad.mit.edu/cgi-bin/cancer/datasets.cgi.
CNS data set
CNS is also an Affymetrix high-density oligonucleotide microarray data set containing 42 samples distributed over 5 different types of tumors of the central nervous system (CNS): 10 medulloblastomas (MD), 10 malignant gliomas (MGlio), 10 atypical teratoid/rhabdoid tumors (Rhab), 8 primitive neuro-ectodermal tumors (PNET), and 4 human cerebella tumors (Ncer). For this data set each sample is represented by 7129 genes. This data set is available at http://www.broad.mit.edu/cgi-bin/cancer/datasets.cgi.
Lung Cancer data set
In this Affymetrix high-density oligonucleotide array, we have 203 samples in 12600 dimensions. There are 5 classes: 139 lung adenocarcinomas (Adeno), 21 squamous cell lung carcinomas (SQ), 20 pulmonary carcinoids (COID), 6 small-cell lung cancer (SCLC), and 17 normal lung specimens (Normal). This data set can be obtained from http://www.pnas.org/content/suppl/2001/11/13/191502998.DC1/DatasetA_12600gene.xls.
Preprocessing
For the Leukemia and CNS data sets, in the preprocessing step the gene expression values less than 100 are raised to 100 and gene expression values greater than 16000 are set to 16000. All gene expression values are then subjected to a base 10 logarithmic transformation. After that, the distribution of gene expression values in each sample is adjusted to zero mean and unit variance. For the SRBCT data set, we do not make any change to the gene expression values as that had already been preprocessed in the original data source [21]. For the Lung Cancer data set, we use the same preprocessed data as used in the original paper [24] without doing any additional preprocessing. For these four data sets, we adopt the same data preprocessing protocols as used in a previous study [1], [15]. In this study, the analysis is conducted using the R environment [87].
Group Marker Index (GMI)
Our main objective is to develop a gene evaluation index, which we call “Group Marker Index (GMI)”, to select biologically significant genes for both two- and multiple-class discrimination problems. GMI builds on the Gene Dominant/Dormant Index (GDI) which was proposed in our earlier study. GDI is a gene evaluation index to select two specific types of genes, i.e., dominant and dormant genes defined in our previous study [1]. In a multiple-class microarray data set, a dominant gene has high gene expression values only in the samples from one specific class and low gene expression values in the samples of the remaining classes. Contrary to the dominant gene, a dormant gene has low gene expression values in only one specific class and high gene expression values in the rest of the classes. GMI is a further refinement of GDI in two aspects: First, irrespective of the number of classes, GDI will only evaluate two levels of discrimination (i.e., dominant and dormant conditions). GMI, on the other hand, will evaluate K-1 levels of discrimination where there are K classes in a pooled microarray experiment. Second, GDI uses the same equation of signal-to-noise ratio (SNR) to evaluate genes, which makes GDI sensitive to outliers, if any. We have avoided these deficiencies by utilizing a simple yet novel intuitive concept, referred to as Between-Class-Transition (BCT), as a further refinement to help identify overlapping classes.
Between-Class-Transition (BCT)
Let 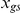 be the gene expression value for the g
th gene in the s
th sample, g = 1, 2, …, G; s = 1, 2, …, S. The s
th sample is associated with a class label
be the gene expression value for the g
th gene in the s
th sample, g = 1, 2, …, G; s = 1, 2, …, S. The s
th sample is associated with a class label 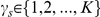 . For each gene g, g = 1, 2, …, G, we sort the gene expression values of the S samples in descending order. In the sorted sequence, each gene expression value is associated with the class label of the corresponding sample. In this sorted sequence, if the class labels of two successive samples (gene expression values) are different, we count it as a between-class-transition (BCT). In this way, for each gene, we find the total number of BCTs.
. For each gene g, g = 1, 2, …, G, we sort the gene expression values of the S samples in descending order. In the sorted sequence, each gene expression value is associated with the class label of the corresponding sample. In this sorted sequence, if the class labels of two successive samples (gene expression values) are different, we count it as a between-class-transition (BCT). In this way, for each gene, we find the total number of BCTs.
Illustration of BCT
We now illustrate BCT using a simple synthetic data set. Consider the gene expression values of a gene for 10 samples divided into two classes (first five samples belong to class 1 and the rest belong to class 2) as depicted in the left panel of Fig. S6. The sorted gene values are depicted in the right panel of Fig. S6. Here we have projected the samples on a vertical line and represented the two classes using two different symbols. In the right panel, the fourth sample from class 1 is sandwiched between two samples from class 2 and this adds two to the BCT count. In this way, the total number of BCTs is 5. An ideal marker gene in a two-class problem will have no outlier sample in either class, and the discrimination between the two classes will be very easy. In such a case the BCT value is only one (for the transition from one class to the other). In all other cases, where there are some outlier samples with gene expression values that overlap with the gene expression values of the samples from the other class, the BCT value will be larger than one. Note that, there could be BCTs even when there is no outlier in the true sense of the word, but the classes have overlap. With an increase in the number of overlapped samples, the BCT value will increase. Therefore, we can use the number of BCTs to help identify overlapped samples, and thus improve marker gene identification.
Consider a two-class problem, as an example. Suppose for a gene g, the samples from each class form compact clusters and the clusters are well separated except for the gene expression value of just one sample in one of the two classes, which is mixed with samples from the other class. In this case, the effect of the outlier on the standard deviation will depend on its location; in other words, how far the outlier is from the mean of the gene expression values among samples in that class. Thus, the effect of just one outlier (mixed sample) on the standard deviation could be moderate to severe. However, this is not the case with BCTs because the number of BCTs will not depend on the location of the outliers in the other class.
Computation of GMI
For easy understanding, Fig. 4 depicts the steps involved in the computation of GMI, which are explained next.
Figure 4. Steps involved to compute GMI and to find the list of group specific genes for each level of discrimination.

Normalization
The gene expression values of each gene are normalized in the range from 0 to 1 across samples. This step preserves the richness in the original gene expression values for each gene among the samples and helps us to easily visualize the distribution of gene expression values for the discriminatory genes.
Computation of the mean value of each class for each gene
For each gene, the mean of the gene expression values in each class is calculated. Let the mean for gene g in class k be μgk.
Sorting of the mean values and defining of K-1 levels of discrimination
For notational simplicity, to explain the computation of the GMI for gene g, we ignore the index g. We sort μk, k = 1, 2, …, K (K = number of classes) in descending order. Let the sorted mean values be μk
(s); k = 1, 2, …, K. Suppose 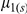 is the mean for class C1
(s). This means that the gene under consideration is the most highly expressed in class C1
(s). We shall call a gene level-n discriminatory gene, if it can discriminate between two groups of classes where one group has n classes in it and the other group has the remaining (K-n) classes. Moreover, for a level-n discriminatory gene, it is highly expressed in the group with n classes while in the remaining K-n classes, this gene is poorly expressed or unexpressed. Note that, if there are K classes, we can define (K-1) levels of discrimination.
is the mean for class C1
(s). This means that the gene under consideration is the most highly expressed in class C1
(s). We shall call a gene level-n discriminatory gene, if it can discriminate between two groups of classes where one group has n classes in it and the other group has the remaining (K-n) classes. Moreover, for a level-n discriminatory gene, it is highly expressed in the group with n classes while in the remaining K-n classes, this gene is poorly expressed or unexpressed. Note that, if there are K classes, we can define (K-1) levels of discrimination.
Computation of GMI for each level of discrimination
For a good marker gene with clear level-n discrimination, there are n classes that have high gene expression values and all other classes have low gene expression values. Therefore, we define the set of classes with high gene expression values as “upper group” and the set of remaining classes as “lower group”. For a good marker gene with level-n discrimination the upper and lower groups should be well separated and the overlap between the groups should be low. For level-n discrimination for gene g, first we focus on the separation between upper and lower groups. We use the difference between the mean values of classes Cn
(s) and Cn
+1(s), i.e., μSep
= μn
(s) – μn
+1(s). This is the closest separation between a class in the upper group and a class in the lower group. We take this as the separation between the two groups of classes. Note that, we are not using the group mean. Next we define a measure of overlap. For this we find the number of BCTs between these two groups. To consider the effect of sample size, we use a weight parameter, NS, to normalize the BCT value. Suppose N
up and N
low represent the total number of samples in upper and lower groups and let NS = Min{N
up, N
low}. Then a measure of overlap between the two groups can be defined as 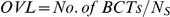 . The GMI value for level-n discrimination for gene g is then defined as the ratio of μSep and OVL :
. The GMI value for level-n discrimination for gene g is then defined as the ratio of μSep and OVL :
| (1) |
A high value of GMI will indicate good separation with low overlap between the two groups.
Finding a list of group specific genes for each level of discrimination
After calculating the GMI values of K-1 levels of discrimination for all genes, a list of group specific genes for each level can be obtained as follows. Every gene has K-1 GMI values. Clearly, genes with higher GMIn values have better level-n discriminating power between upper and lower groups. Now for a given discrimination level-n, we sort all genes in descending order of GMIn values. The sorted values may be denoted as GMIn(s). A smaller rank indicates a larger GMIn value and hence a better level-n discriminating power of the gene. From the top of the list of sorted level-n genes, we can select a set of genes that is likely to be biologically interesting and is expected to be useful for level-n discrimination. Note that, the upper and lower groups associated with different level-n genes could be completely different. At this point, we would like to emphasize that designing of classifier or diagnostic prediction system is not the primary objective of this study. The main objective is to find genes that can discriminate between two sets of diseases, not necessarily one disease verses other. We also emphasize that the proposed scheme neither uses pooled mean of a group of classes nor uses the pooled standard deviation of a set of classes and hence is free from the problems of SNR type indexes [1].
Illustration of GMI Computation
To understand the computation of the GMI value at each level of discrimination for a given gene, we have generated a synthetic example, which is composed of five classes of samples. As shown in Fig. 5, there are five classes of samples and there are ten samples in each class. The samples from different classes are labeled with different colors. Since there are K = 5 classes, we evaluate the gene for 4 possible levels of discrimination. In Fig. 5, panel a to panel d, represent, respectively, the computation of level-1 to level-4 discrimination of this gene. In panel a, the level-1 discrimination is evaluated between the third class and the fourth class because they are ranked first and second based on the descending order of the mean of the gene expression values in the five classes. These two highest ranked means are used to calculate μSep and this separation is indicated by two horizontal dashed lines in green and blue. For an easy computation of BCTs, in all four panels, the upper and lower groups are represented using filled in and empty symbols, respectively. Similarly, for panels b to d. For example, in panel b, for level-2 discrimination, μSep is computed using the mean values of class four and class five. From these figures, it is clear that this synthetic gene is a good level-2 gene because from panel b we find that two groups are well separated and the BCT is just 1. This is also revealed by the GMI values of 0.150, 9.537, 0.287, and 1.925 for level-1 to level-4, respectively.
Figure 5. A 5-class synthetic example to illustrate computation of GMI.

There are four levels of discrimination in the 5-class synthetic data set. Panels (a) to (d) depict the computation of GMI values at each level of discrimination. The dotted lines in each panel indicate the two mean values used for GMI computation in each level of discrimination. All filled samples in each panel indicate the upper group samples. The remaining open samples in each panel indicate the lower group samples.
Gene selection and evaluation of statistical significance
To evaluate the statistical significance of group specific genes identified by GMI, we perform a permutation procedure to obtain the corresponding p- and q-values. This procedure is similar to the method used in our previous study [1]. The necessary steps are summarized below. Let G be the total number of genes and S be the total number of samples.
Step 1. Gene selection
Step 1.1 Repeated random splitting. Given a microarray data set D with K classes (xgs is the gene expression value of gene g in sample s; 1≤g ≤G, 1≤s≤S) and with class labels (γs, 1≤s≤S), we randomly select 2/3rd samples from each class as the training set TR(r) (r denotes the r th random selection of samples, 1≤r≤R).
Step 1.2 Computation of GMI and preliminary gene selection. For each training set TR(r), we compute the GMI values for different levels of discrimination GMIng(r) (1≤n≤K-1) for each gene g. Simultaneously, we use an independent indicator Fng(r) to denote whether the gene g is included in the list of top N1 genes (N1 genes ranked in descending order of GMIng(r) values); Fng(r) = 1 represents “true” and Fng(r) = 0 represents “false”.
Step 1.3 Gene selection. After R = 100 times of the random selection of samples, we average the GMI values of different levels of discrimination, GMIng(r), 1≤r≤R, for each gene g as GMIng(ave) and sum up the Fng(r) values for each gene g as Fng(sum). Note that, Fng(sum) is the number of times (i.e., frequency) the gene g is selected as one of the top N1 genes in R experiments. For each level of discrimination, we select the top N2 genes with the highest frequencies, Fng(sum). In this study, we set both N1 and N2 = 10.
Step 2. Permutation
We randomly permute the class labels γs for B times. In the b th permutation (1≤b≤B), we randomly select 2/3rd samples from each class as the training set TR(r)(b) for R = 100 times. For each training set TR(r)(b), we compute GMIng(r) (b) for different levels of discrimination for gene g using the permuted class labels γs (b). Next, we average these new GMI values of different levels of discrimination GMIng(r)(b) for each gene g as GMIng(ave)(b). These GMIng(ave)(b) are used for the calculation of p- and q-values.
Step 3. Calculation of p-values
The p-value of the observed averaged GMI value, GMIng(ave), for a particular level of discrimination, of a gene g is
| (2) |
Where I(.) is an indicator function that takes the value one when true, and zero otherwise.
Step 4. Calculation of q-values
To account for the multiple tests being performed in the G genes, the q-value of the observed averaged GMIng(ave) is calculated as
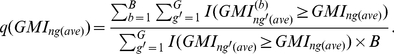 |
(3) |
In this study, we have performed this permutation test with B = 200 for all data sets.
Comparison with other method
In order to demonstrate the effectiveness of our GMI method in identifying group specific genes, we propose a scheme based on the template-based method (TBM), which is similar to the method used in a previous work [19] for comparison. In this study, we used Pearson's correlation coefficient to evaluate the relation between gene expression values and pre-assigned templates. The detailed steps of gene selection in the proposed TBM are described below:
TBM Step 1: Repeated random splitting
For a fair comparison with GMI, the repeated random splitting scheme is also used here. For a given microarray data set D with K classes (xgs is the gene expression value of gene g in sample s; 1≤g≤G, 1≤s≤S) with class labels (γs, 1≤s≤S), we randomly select 2/3rd samples from each class as the training set TR(r) (as earlier, r denotes the r th random selection of samples, 1≤r≤R).
TBM Step 2: Normalization
For each training set TR(r), the gene expression values of every gene are normalized across samples to [0, 1].
TBM Step 3: Identification of group specific genes for level-n discrimination
For every gene in the training set TR(r), K-1 levels of discrimination are defined as in the previous section. To find group specific marker genes for level-n discrimination, the following steps are followed:
TBM Step 3.1 Creation of template T0(r).We create a template T0(r){0, 0, 0, … , 0}; the length of T0(r) is equal to the number of samples in training set TR(r). Initially, every value in T0(r) is set to zero.
TBM Step 3.2 Creation of template Tn(m). For a given level-n, there could be many combinations (subsets) of classes for the upper group. We denote the appropriate template for the m th combination (for level-n discrimination) as Tn(m)(r) and generate it from T0(r) as follows. If the s th sample of gene g belongs to the upper group (i.e., in the m th combination of classes) then the s th value of T0(r) is set to 1. This modified T0(r) is the template Tn(m)(r).
TBM Step 3.3 Computation of Pearson's correlation coefficient. Note that, for level-n, m
th combination, and sample set TR(r), the Tn(m)(r) is fixed. Now we compute the Pearson's correlation coefficient for every gene g, with the template Tn(m)(r) and denote it as Pn(m)(r)(g). For level-n we have 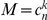 combinations. Let
combinations. Let 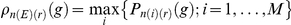 . Now we sort the G correlation values
. Now we sort the G correlation values 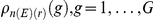 in descending order and create the list of top N1 genes (along with the associated upper groups) for level-n as Ln
(r). Thus, a typical element of the list will have two components: the upper group (E) and the gene (g).
in descending order and create the list of top N1 genes (along with the associated upper groups) for level-n as Ln
(r). Thus, a typical element of the list will have two components: the upper group (E) and the gene (g).
TBM Step 4: Gene selection
Steps 1 through 3 are repeated for r = 1 to R times (here R = 100) resulting in R lists of top N1 genes, Ln(r); r = 1, …, R. Let Fn(g, m) be the number of times (frequency) with which the gene g, associated with upper group m, appears in the R lists. This results in a list where every gene has associated with it just one frequency and one upper group. Now, we sort this list in descending order based on the frequencies and select the top N2 genes from that list. Note that, each of these N2 genes will have an associated upper group. Here N1 = N2 = 10 as used with GMI.
Supporting Information
Scatter-plots of the top most gene of each level in the Leukemia data set.
(PDF)
Scatter-plots of the top most gene of each level in the CNS data set.
(PDF)
Scatter-plots of the top most gene of each level in the Lung Cancer data set.
(PDF)
The modified figure of B cell receptor signaling pathway.
(PDF)
The distributions of ANOVA p-values for four data sets.
(PDF)
Illustration of between-class-transition (BCT).
(PDF)
Summary of top 10 genes of each level selected by GMI in the Leukemia data set.
(PDF)
Summary of top 10 genes of each level selected by GMI in the CNS data set.
(PDF)
Summary of top 10 genes of each level selected by GMI in the Lung Cancer data set.
(PDF)
The comparison of top 10 level-2 genes selected by GMI and TBM in the CNS data set.
(PDF)
The comparison of top 10 level-3 genes selected by GMI and TBM in the CNS data set.
(PDF)
The comparison of top 10 level-2 genes selected by GMI and TBM in the Leukemia data set.
(PDF)
The comparison of top 10 level-2 genes selected by GMI and TBM in the Lung Cancer data set.
(PDF)
The comparison of top 10 level-3 genes selected by GMI and TBM in the Lung Cancer data set.
(PDF)
The full summary table of the identified pathways related to the level 2 discriminatory genes in the Leukemia data set.
(PDF)
The full summary table of the identified pathways related to the level 2 discriminatory genes in the Lung cancer data set (Part I).
(PDF)
The full summary table of the identified pathways related to the level 2 discriminatory genes in the Lung cancer data set (Part II).
(PDF)
The level-2 genes selected only by GMI and the level-2 genes selected only by TBM in the Lung Cancer data set.
(PDF)
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work is supported by National Science Council (NSC) (NSC100-2221-E-010-011 and NSC99-2627-B-010-009). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Tsai YS, Lin CT, Tseng GC, Chung IF, Pal NR. Discovery of dominant and dormant genes from expression data using a novel generalization of SNR for multi-class problems. BMC Bioinformatics. 2008;9:425. doi: 10.1186/1471-2105-9-425. doi: 10.1186/1471-2105-9-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 3.Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc. 2002;97:77–87. doi: 10.1198/016214502753479248. [Google Scholar]
- 4.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach Learn. 2002;46:389–422. [Google Scholar]
- 5.Pal NR, Aguan K, Sharma A, Amari SI. Discovering biomarkers from gene expression data for predicting cancer subgroups using neural networks and relational fuzzy clustering. BMC Bioinformatics. 2007;8:5. doi: 10.1186/1471-2105-8-5. doi: 10.1186/1471-2105-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saeys Y, Inza I, Larrañaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23:2507–2517. doi: 10.1093/bioinformatics/btm344. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 7.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Opgen-Rhein R, Strimmer K. Accurate ranking of differentially expressed genes by a distribution-free shrinkage approach. Stat Appl Genet Mol Biol. 2007;6:9. doi: 10.2202/1544-6115.1252. doi: 10.2202/1544-6115.1252. [DOI] [PubMed] [Google Scholar]
- 9.Zuber V, Strimmer K. Gene ranking and biomarker discovery under correlation. Bioinformatics. 2009;25:2700–2707. doi: 10.1093/bioinformatics/btp460. doi: 10.1093/bioinformatics/btp460. [DOI] [PubMed] [Google Scholar]
- 10.Liu XX, Krishnan A, Mondry A. An Entropy-based gene selection method for cancer classification using microarray data. BMC Bioinformatics. 2005;6:76. doi: 10.1186/1471-2105-6-76. doi: 10.1186/1471-2105-6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huerta EB, Duval B, Hao JK. Fuzzy logic for elimination of redundant information of microarray data. Genomics Proteomics Bioinformatics. 2008;6(2):61–73. doi: 10.1016/S1672-0229(08)60021-2. doi: 10.1016/S1672-0229(08)60021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Weinberg CR, Darden TA, Pedersen LG. Gene selection for sample classification based on gene expression data: study of sensitivity to choice of parameters of the GA/KNN method. Bioinformatics. 2001;17:1131–1142. doi: 10.1093/bioinformatics/17.12.1131. doi: 10.1093/bioinformatics/17.12.1131. [DOI] [PubMed] [Google Scholar]
- 13.Ooi CH, Tan P. Genetic algorithms applied to multi-class prediction for the analysis of gene expression data. Bioinformatics. 2003;19:37–44. doi: 10.1093/bioinformatics/19.1.37. doi: 10.1093/bioinformatics/19.1.37. [DOI] [PubMed] [Google Scholar]
- 14.Jirapech-Umpai T, Aitken S. Feature selection and classification for microarray data analysis: evolutionary methods for identifying predictive genes. BMC Bioinformatics. 2005;6:148. doi: 10.1186/1471-2105-6-148. doi: 10.1186/1471-2105-6-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niijima S, Kuhara S. Recursive gene selection based on maximum margin criterion: a comparison with SVM-RFE. BMC Bioinformatics. 2006;7:543. doi: 10.1186/1471-2105-7-543. doi: 10.1186/1471-2105-7-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Tuck DP. MSVM-RFE: extensions of SVM-RFE for multiclass gene selection on DNA microarray data. Bioinformatics. 2007;23:1106–1114. doi: 10.1093/bioinformatics/btm036. doi: 10.1093/bioinformatics/btm036. [DOI] [PubMed] [Google Scholar]
- 17.Yousef M, Jung S, Showe LC, Showe MK. Recursive Cluster Elimination (RCE) for classification and feature selection from gene expression data. BMC Bioinformatics. 2007;8:144. doi: 10.1186/1471-2105-8-144. doi: 10.1186/1471-2105-8-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yousef M, Ketany M, Manevitz L, Showe LC, Showe MK. Classification and biomarker identification using gene network modules and support vector machines. BMC Bioinformatics. 2009;10:337. doi: 10.1186/1471-2105-10-337. doi: 10.1186/1471-2105-10-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavlidis P, Noble WS. Analysis of strain and regional variation in gene expression in mouse brain. Genome Biol. 2001;2:42. doi: 10.1186/gb-2001-2-10-research0042. doi: 10.1186/gb-2001-2-10-research0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hulshizer R, Blalock EM. Post hoc pattern matching: assigning significance to statistically defined expression patterns in single channel microarray data. BMC Bioinformatics. 2007;8:240. doi: 10.1186/1471-2105-8-240. doi: 10.1186/1471-2105-8-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan J, Wei JS, Ringnér M, Saal LH, Ladanyi M, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 23.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 24.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan J, Wei JS, Ringnér M, Saal LH, Ladanyi M, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baird K, Davis S, Antonescu CR, Harper UL, Walker RL, et al. Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer Res. 2005;65:9226–9235. doi: 10.1158/0008-5472.CAN-05-1699. doi: 10.1158/0008-5472.CAN-05-1699. [DOI] [PubMed] [Google Scholar]
- 27.Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, et al. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med. 2004;10:175–181. doi: 10.1038/nm966. doi: 10.1038/nm966. [DOI] [PubMed] [Google Scholar]
- 28.Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006;66:6936–6946. doi: 10.1158/0008-5472.CAN-05-4578. doi: 10.1158/0008-5472.CAN-05-457. [DOI] [PubMed] [Google Scholar]
- 29.Taylor JG VI, Cheuk AT, Tsang PS, Chung JY, Song YK, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119:3395–3407. doi: 10.1172/JCI39703. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerisano V, Aalto Y, Perdichizzi S, Bernard G, Manara MC, et al. Molecular mechanisms of CD99-induced caspase-independent cell death and cell-cell adhesion in Ewing's sarcoma cells: actin and zyxin as key intracellular mediators. Oncogene. 2004;23:5664–5674. doi: 10.1038/sj.onc.1207741. doi: 10.1038/sj.onc.1207741. [DOI] [PubMed] [Google Scholar]
- 31.Rocchi A, Manara MC, Sciandra M, Zambelli D, Nardi F, et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J Clin Invest. 2010;120:668–680. doi: 10.1172/JCI36667. doi: 10.1172/JCI36667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scotlandi K, Baldini N, Cerisano V, Manara MC, Benini S, et al. CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res. 2000;60:5134–5142. [PubMed] [Google Scholar]
- 33.Salagierski M, Verhaegh GW, Jannink SA, Smit FP, Hessels D, et al. Differential expression of PCA3 and its overlapping PRUNE2 transcript in prostate cancer. Prostrate. 2009;70:70–78. doi: 10.1002/pros.21040. doi: 10.1002/pros.21040. [DOI] [PubMed] [Google Scholar]
- 34.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–958. doi: 10.1126/science.285.5429.895. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Liu YE, Greene J, Sheng S, Fuchs A, et al. Inhibition of tumor growth and metastasis of human breast cancer cells transfected with tissue inhibitor of metalloproteinase 4. Oncogene. 1997;14:2767–2774. doi: 10.1038/sj.onc.1201245. [DOI] [PubMed] [Google Scholar]
- 36.Kostyukova AS, Rapp BA, Choy A, Greenfield NJ, Hitchcock-DeGregori SE. Structural requirements of tropomodulin for tropomyosin binding and actin filament capping. Biochemistry. 2005;44:4905–4910. doi: 10.1021/bi047468p. doi:10.1021/bi047468. [DOI] [PubMed] [Google Scholar]
- 37.Dilley WG, Kalyanaraman S, Verma S, Cobb JP, Laramie JM, et al. Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol Cancer. 2005;4:9. doi: 10.1186/1476-4598-4-9. doi: 10.1186/1476-4598-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bishopric NH, Jayasena V, Webster KA. Positive regulation of the skeletal alpha-actin gene by Fos and Jun in cardiac myocytes. J Biol Chem. 1992;267:25535–25540. [PubMed] [Google Scholar]
- 39.Seo S, Lim JW, Yellajoshyula D, Chang LW, Kroll KL. Neurogenin and NeuroD direct transcriptional targets and their regulatory enhancers. EMBO J. 2007;26:5093–5108. doi: 10.1038/sj.emboj.7601923. doi: 10.1038/sj.emboj.7601923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martoglio AM, Tom BD, Starkey M, Corps AN, Charnock-Jones DS, et al. Changes in tumorigenesis- and angiogenesis-related gene transcript abundance profiles in ovarian cancer detected by tailored high density cDNA arrays. Mol Med. 2000;6:750–765. [PMC free article] [PubMed] [Google Scholar]
- 41.Yap CT, Simpson TI, Pratt T, Price DJ, Maciver SK. The motility of glioblastoma tumour cells is modulated by intracellular cofilin expression in a concentration-dependent manner. Cell Motil Cytoskeleton. 2005;60:153–165. doi: 10.1002/cm.20053. doi: 10.1002/cm.20053. [DOI] [PubMed] [Google Scholar]
- 42.Campbell DH, deFazio A, Sutherland RL, Daly RJ. Expression and tyrosine phosphorylation of EMS1 in human breast cancer cell lines. Int J Cancer. 1996;68:485–92. doi: 10.1002/(SICI)1097-0215(19961115)68:4<485::AID-IJC14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 43.Patel AM, Incognito LS, Schechter GL, Wasilenko WJ, Somers KD. Amplification and expression of EMS-1 (cortactin) in head and neck squamous cell carcinoma cell lines. Oncogene. 1996;12:31–35. [PubMed] [Google Scholar]
- 44.Ishii H, Baffa R, Numata SI, Murakumo Y, Rattan S, et al. The FEZ1 gene at chromosome 8p22 encodes a leucine-zipper protein, and its expression is altered in multiple human tumors. Proc Natl Acad Sci USA. 1999;96:3928–3933. doi: 10.1073/pnas.96.7.3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Q, Jones TI, Tang VW, Brieher WM, Jones PL. Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J Cell Sci. 2010;123:1116–1123. doi: 10.1242/jcs.058958. doi: 10.1242/jcs.058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerlitz G, Bustin M. Efficient cell migration requires global chromatin condensation. J Cell Sci. 2010;123:2207–2217. doi: 10.1242/jcs.058271. doi: 10.1242/jcs.058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bustin M. Regulation of DNA-Dependent Activities by the Functional Motifs of the High Mobility-Group Chromosomal Proteins. Mol Cell Biol. 1999;19(8):5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linial M, Miller K, Scheller RH. VAT-1: an abundant membrane protein from Torpedo cholinergic synaptic vesicles. Neuron. 1989;2:1265–1273. doi: 10.1016/0896-6273(89)90311-5. [DOI] [PubMed] [Google Scholar]
- 49.Han C, Chen T, Yang M, Li N, Liu H, et al. Human SCAMP5, a Novel Secretory Carrier Membrane Protein, Facilitates Calcium-Triggered Cytokine Secretion by Interaction with SNARE Machinery. J Immunol. 2009;182:2986–2996. doi: 10.4049/jimmunol.0802002. doi: 10.4049/jimmunol.0802002. [DOI] [PubMed] [Google Scholar]
- 50.Depaz IM, de Las Heras R, Kroon PA, Wilce PA. Changes in neuronal protein 22 expression and cytoskeletal association in the alcohol-dependent and withdrawn rat brain. J Neurosci Res. 2005;81:253–60. doi: 10.1002/jnr.20563. doi: 10.1002/jnr.20563. [DOI] [PubMed] [Google Scholar]
- 51.Gu Y, Ihara Y. Evidence that collapsin response mediator protein-2 is involved in the dynamics of microtubules. J Biol Chem. 2000;275:17917–20. doi: 10.1074/jbc.C000179200. doi: 10.1074/jbc.C000179200. [DOI] [PubMed] [Google Scholar]
- 52.Lan MS, Breslin MB. Structure, expression, and biological function of INSM1 transcription factor in neuroendocrine differentiation. FASEB J. 2009;23(7):2024–2033. doi: 10.1096/fj.08-125971. doi: 10.1096/fj.08-125971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu WD, Wang HW, Muguira M, Breslin MB, Lan MS. INSM1 functions as a transcriptional repressor of the neuroD/β2 gene through the recruitment of cyclin D1 and histone deacetylases. Biochem J. 2006;397(Pt 1):169–177. doi: 10.1042/BJ20051669. doi: 10.1042/BJ20051669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ren WZ, Ng GY, Wang RX, Wu PH, O'Dowd BF, et al. The identification of NP25: a novel protein that is differentially expressed by neuronal subpopulations. Brain Res Mol Brain Res. 1994;22:173–185. doi: 10.1016/0169-328x(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 55.Yamashita N, Uchida Y, Ohshima T, Hirai S, Nakamura F, et al. Collapsin response mediator protein 1 mediates reelin signaling in cortical neuronal migration. J Neuroscience. 2006;26(51):13357–13362. doi: 10.1523/JNEUROSCI.4276-06.2006. doi: 10.1523/JNEUROSCI.4276-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atz ME, Rollins B, Vawter MP. NCAM1 association study of bipolar disorder and schizophrenia: polymorphisms and alternatively spliced isoforms lead to similarities and differences. Psychiatr Genet. 2007;17(2):55–67. doi: 10.1097/YPG.0b013e328012d850. doi: 10.1097/YPG.0b013e328012d850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernández-Chacón R, Südhof TC. Novel SCAMPs Lacking NPF Repeats: Ubiquitous and Synaptic Vesicle-Specific Forms Implicate SCAMPs in Multiple Membrane-Trafficking Functions. J Neurosci. 2000;20(21):7941–7950. doi: 10.1523/JNEUROSCI.20-21-07941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe H, Mizutani T, Haraguch T, Yamamichi N, Minoguchi S, et al. SWI/SNF complex is essential for NRSF-mediated suppression of neuronal genes in human nonsmall cell lung carcinoma cell lines. Oncogene. 2006;25:470–479. doi: 10.1038/sj.onc.1209068. doi: 10.1038/sj.onc.1209068. [DOI] [PubMed] [Google Scholar]
- 59.Tian T, Hao J, Xu A, Hao J, Luo C, et al. Determination of metastasis-associated proteins in non-small-cell lung cancer by comparative proteomic analysis. Cancer Sci. 2007;98:1265–74. doi: 10.1111/j.1349-7006.2007.00514.x. doi: 10.1111/j.1349-7006.2007.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, Fogg DK, Waisman DM. RNA interference-mediated silencing of the S100A10 gene attenuates plasmin generation and invasiveness of Colo 222 colorectal cancer cells. J Biol Chem. 2004;279:2053–2062. doi: 10.1074/jbc.M310357200. doi: 10.1074/jbc.M310357200. [DOI] [PubMed] [Google Scholar]
- 61.Martins L, Matsuo SE, Ebina KN, Kulcsar MA, Friguglietti CU, et al. Galectin-3 messenger ribonucleic acid and protein are expressed in benign thyroid tumors. J Clin Endocrinol Metab. 2002;87:4806–10. doi: 10.1210/jc.2002-020094. doi: 10.1210/jc.2002-020094. [DOI] [PubMed] [Google Scholar]
- 62.Yoshimura A, Gemma A, Hosoya Y, Komaki E, Hosomi Y, et al. Increased expression of the LGALS3 (galactin 3) gene is human non-small cell lung cancer. Genes Chromosomes Cancer. 2003;37:159–64. doi: 10.1002/gcc.10205. doi: 10.1002/gcc.10205. [DOI] [PubMed] [Google Scholar]
- 63.McHugh BJ, Buttery R, Lad Y, Banks S, Haslett C, et al. Integrin activation by Fam38A uses a novel mechanism of R-Ras targeting to the endoplasmic reticulum. J Cell Sci. 2010;123:51–61. doi: 10.1242/jcs.056424. doi: 10.1242/jcs.056424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–357. doi: 10.1093/nar/gkj102. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 68.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Alphen RJ, Wiemer EAC, Burger H, Eskens ALM. The spliceosome as target for anticancer treatment. Br J Cancer. 2009;100(2):228–232. doi: 10.1038/sj.bjc.6604801. doi: 10.1038/sj.bjc.6604801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nahar R, Müschen M. Pre-B cell receptor signaling in acute lymphoblastic leukemia. Cell Cycle. 2009;8(23):3874–3877. doi: 10.4161/cc.8.23.10035. doi: 10.4161/cc.8.23.10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Downing JR. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol. 1999;106(2):296–308. doi: 10.1046/j.1365-2141.1999.01377.x. doi: 10.1046/j.1365-2141.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 72.Sun L, Heerema N, Crotty L, Wu X, Navara C et al. Expression of dominant-negative and mutant isoforms of the antileukemic transcription factor Ikaros in infant acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1999;96(2):680–685. doi: 10.1073/pnas.96.2.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mengubas K, Jabbar S, Nye K, Wilkes S, Hoffbrand AV, et al. Inactivation of calcium ion-regulating inositol polyphosphate 2nd-messengers is impaired in subpopulations of human leukemia-cells. Leukemia. 1994;8(10):1718–1725. [PubMed] [Google Scholar]
- 74.Fiancette R, Vincent c, Donnard M, Bordessoule d, Turlure P, et al. Genes encoding multiple forms of phospholipase A2 are expressed in immature forms of human leukemic blasts. Leukemia. 2009;23:1196–1199. doi: 10.1038/leu.2009.36. doi: 10.1038/leu.2009.36. [DOI] [PubMed] [Google Scholar]
- 75.Agatha G, Häfer R, Zintl F. Fatty acid composition of lymphocyte membrane phospholipids in children with acute leukemia. Cancer Lett. 2001;173(2):139–144. doi: 10.1016/s0304-3835(01)00674-7. [DOI] [PubMed] [Google Scholar]
- 76.Chiarini F, Falà F, Tazzari PL, Ricci F, Astolfi A, et al. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of Rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009;69:3520–3528. doi: 10.1158/0008-5472.CAN-08-4884. doi: 10.1158/0008-5472.CAN-08-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8:380–390. doi: 10.1038/nri2304. doi: 10.1038/nri2304. [DOI] [PubMed] [Google Scholar]
- 78.Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, et al. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood. 2005;106(12):3898–3906. doi: 10.1182/blood-2005-01-0355. doi: 10.1182/blood-2005-01-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tavor S, Eisenbach M, Jacob-Hirsch J, Golan T, Petit I, et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia. 2008;22:2151–2158. doi: 10.1038/leu.2008.238. doi: 10.1038/leu.2008.238. [DOI] [PubMed] [Google Scholar]
- 80.Bruserud Ø, Gjertsen BT, Ulvestad E. Expression of Fcε-receptors by human acute myelogenous leukemia (AML) blasts: studies of high- and low- (CD23) affinity receptor expression and the effects of IgE-mediated receptor ligation on functional AML blast characteristics. Leuk Res. 2002;26(5):515–521. doi: 10.1016/s0145-2126(01)00165-5. doi: 10.1016/S0145-2126(01)00165-5. [DOI] [PubMed] [Google Scholar]
- 81.Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, et al. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11:886–891. doi: 10.1038/nm1274. doi: 10.1038/nm1274. [DOI] [PubMed] [Google Scholar]
- 82.Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–449. doi: 10.1126/science.1088485. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- 83.Kuroda K, Takenoyama M, Baba T, Shigematsu Y, Shiota H, et al. Identification of ribosomal protein L19 as a novel tumor antigen recognized by autologous cytotoxic T lymphocytes in lung adenocarcinoma. Cancer Sci. 2009;101(1):46–55. doi: 10.1111/j.1349-7006.2009.01351.x. doi: 10.1111/j.1349-7006.2009.01351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nadano D, Notsu T, Matsuda T, Sato TA. A human gene encoding a protein homologous to ribosomal protein L39 is normally expressed in the testis and derepressed in multiple cancer cells. Biochim Biophys Acta. 2002;1577(3):430–436. doi: 10.1016/s0167-4781(02)00445-1. doi: 10.1016/S0167-4781(02)00445-1. [DOI] [PubMed] [Google Scholar]
- 85.Ocak S, Yamashita H, Udyavar AR, Miller AN, Gonzalez AL, et al. DNA copy number aberrations in small-cell lung cancer reveal activation of the focal adhesion pathway. Oncogene. 2010;29:6331–6342. doi: 10.1038/onc.2010.362. doi: 10.1038/onc.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mukhopadhyay NK, Gordon GJ, Chen CJ, Bueno R, David J, Sugarbaker DJ, et al. Activation of focal adhesion kinase in human lung cancer cells involves multiple and potentially parallel signaling events. J Cell Mol Med. 2007;9(2):387–397. doi: 10.1111/j.1582-4934.2005.tb00364.x. doi: 10.1111/j.1582-4934.2005.tb00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.R Development Core Team. Vienna, Austria: R Foundation for Statistical Computing; 2010. R: A language and environment for statistical computing. ISBN 3-900051-07-0, URL http://www.R-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scatter-plots of the top most gene of each level in the Leukemia data set.
(PDF)
Scatter-plots of the top most gene of each level in the CNS data set.
(PDF)
Scatter-plots of the top most gene of each level in the Lung Cancer data set.
(PDF)
The modified figure of B cell receptor signaling pathway.
(PDF)
The distributions of ANOVA p-values for four data sets.
(PDF)
Illustration of between-class-transition (BCT).
(PDF)
Summary of top 10 genes of each level selected by GMI in the Leukemia data set.
(PDF)
Summary of top 10 genes of each level selected by GMI in the CNS data set.
(PDF)
Summary of top 10 genes of each level selected by GMI in the Lung Cancer data set.
(PDF)
The comparison of top 10 level-2 genes selected by GMI and TBM in the CNS data set.
(PDF)
The comparison of top 10 level-3 genes selected by GMI and TBM in the CNS data set.
(PDF)
The comparison of top 10 level-2 genes selected by GMI and TBM in the Leukemia data set.
(PDF)
The comparison of top 10 level-2 genes selected by GMI and TBM in the Lung Cancer data set.
(PDF)
The comparison of top 10 level-3 genes selected by GMI and TBM in the Lung Cancer data set.
(PDF)
The full summary table of the identified pathways related to the level 2 discriminatory genes in the Leukemia data set.
(PDF)
The full summary table of the identified pathways related to the level 2 discriminatory genes in the Lung cancer data set (Part I).
(PDF)
The full summary table of the identified pathways related to the level 2 discriminatory genes in the Lung cancer data set (Part II).
(PDF)
The level-2 genes selected only by GMI and the level-2 genes selected only by TBM in the Lung Cancer data set.
(PDF)