

Abstract
Low-grade inflammation in adipose tissue and liver has been implicated in obesity-associated insulin resistance and type 2 diabetes. Yet, the contribution of inflammatory cells to the pathogenesis of skeletal muscle insulin resistance remains elusive. In a large cohort of obese human individuals, blood monocyte Fas (CD95) expression correlated with systemic and skeletal muscle insulin resistance. To test a causal role for myeloid cell Fas expression in the development of skeletal muscle insulin resistance, we generated myeloid/haematopoietic cell-specific Fas-depleted mice. Myeloid/haematopoietic Fas deficiency prevented the development of glucose intolerance in high fat-fed mice, in ob/ob mice, and in mice acutely challenged by LPS. In vivo, ex vivo and in vitro studies demonstrated preservation of muscle insulin responsiveness with no effect on adipose tissue or liver. Studies using neutralizing antibodies demonstrated a role for TNFα as mediator between myeloid Fas and skeletal muscle insulin resistance, supported by significant correlations between monocyte Fas expression and circulating TNFα in humans. In conclusion, our results demonstrate an unanticipated crosstalk between myeloid cells and skeletal muscle in the development of obesity-associated insulin resistance.
Keywords: diabetes mellitus, insulin resistance, obesity
Introduction
Obese people are prone to type 2 diabetes, arising from relative insulin deficiency combined with insulin resistance. The latter is defined as impaired ability of insulin to promote glucose uptake into skeletal muscle and adipose tissue as well as to inhibit hepatic glucose production. Since skeletal muscle is the predominant site of insulin-mediated glucose disposal in the postprandial state (DeFronzo & Tripathy, 2009), development of muscular insulin resistance will crucially impact on glucose homeostasis. In fact, skeletal muscle insulin resistance is an early metabolic alteration that frequently precedes type 2 diabetes (DeFronzo & Tripathy, 2009). In obesity, low-grade inflammation frequently develops systemically and in different organs such as adipose tissue, liver or pancreatic islets, and is thought to play a causative role in the pathogenesis of insulin resistance and type 2 diabetes (Donath & Shoelson, 2011; Lazar, 2005; Weisberg et al, 2003; Xu et al, 2003; Yuan et al, 2001). It is characterized by elevated levels of pro-inflammatory cytokines, and by infiltration of these tissues with macrophages and other immune cells (Nishimura et al, 2009; Weisberg et al, 2003; Winer et al, 2009, 2011; Xu et al, 2003). Moreover, although macrophage accumulation was mainly studied in adipose tissue, skeletal muscle may also be infiltrated, a process proposed to contribute to high fat diet (HFD) induced insulin resistance (Olefsky & Glass, 2010). However, the contribution of inflammatory cells including monocytes and macrophages to skeletal muscle insulin resistance remains poorly understood (Mauer et al, 2010; Samokhvalov et al, 2009).
Fas–FasL interaction is regarded as an important action in immune responses. Besides its well-characterized role in the induction of programmed cell death (apoptosis), there is evidence that Fas activation may induce non-apoptotic signalling pathways such as inflammatory cascades (Guo et al, 2005; Peter et al, 2007; Schumann et al, 2007; Wajant et al, 2003). Accordingly, activation of the Fas signalling pathway was shown to induce secretion of pro-inflammatory cytokines like IL-1α, IL-1β, IL-6, IL-8 (keratinocyte chemoattractant (KC)), MCP-1 and TNFα in different cell types such as adipocytes and monocytes (Faouzi et al, 2001; Farley et al, 2008; Imamura et al, 2004; Miwa et al, 1998; Park et al, 2003; Schaub et al, 2003; Wang et al, 2010; Wueest et al, 2010b), rendering it a potential key component of the inflammatory response. We previously found that Fas ablation specifically in adipocytes reduced adipose tissue inflammation and hepatic insulin resistance in high fat-fed mice, highlighting a role for Fas in the disturbed adipocyte–hepatocyte communication observed in obesity (Wueest et al, 2010b).
In the present study, we hypothesized that myeloid cell-expressed Fas impacts on inflammatory pathways and, consequently, on glucose homeostasis. To test this hypothesis, monocytic Fas expression was analysed in a large cohort of obese human individuals (n = 246) and correlated with the parameters insulin sensitivity and glucose metabolism. In addition, glucose metabolism was determined in standard chow-fed, HFD-fed or LPS-injected control and myeloid cell-specific Fas-depleted mice on a C57BL6/J or ob/ob background. Our results propose a thus far unknown Fas-mediated crosstalk between myeloid cells and skeletal muscle contributing to the development of obesity-associated insulin resistance.
Results
Fas expression in circulating monocytes correlates with insulin resistance and type 2 diabetes in obese patients
To unravel whether obesity has an impact on myeloid Fas expression, mRNA levels were determined in circulating monocytes of lean and obese human subjects (body mass index (BMI): 21.4 ± 0.5 kg/m2 in lean vs. 45.9 ± 1.1 kg/m2 in obese subjects, p < 0.0001). Intriguingly, Fasexpression was significantly increased in obese compared to lean subjects (Fig 1A). To further determine if Fas expression in circulating monocytes signifies a more metabolically morbid sub-phenotype of human obesity, monocytic Fas mRNA expression was analysed in obese humans with either normal glucose tolerance (NGT; n = 131) or with type 2 diabetes (n = 115). Basic clinical characteristics of these subjects are provided in Table 1. Fas expression was not different between males and females, but higher in monocytes of obese persons with type 2 diabetes compared to obese, normal glucose tolerant subjects (Fig 1B). Similar to Fas, Fas ligand (FasL) expression was increased in obese persons with type 2 diabetes (supplementary Fig 1). Strikingly, Fas expression in human circulating monocytes positively correlated with HOMA-IR (Fig 1C), a measure of systemic insulin resistance. To gain more insight into potential mechanisms linking monocytic Fas expression and insulin resistance, hyperinsulinaemic-euglycaemic clamp studies were performed. Fas mRNA in circulating monocytes correlated negatively with glucose disposal rate (GDR), a measure mainly reflecting skeletal muscle insulin sensitivity (Fig 1D). Complementary to the cross-sectional study, surgery-induced weight loss, which resulted in significantly improved insulin sensitivity (supplementary Fig 2), also resulted in a significant decline in monocyte Fas mRNA expression (Fig 1E). Clinical characteristics of these subjects are provided in Table 1. Importantly, HOMA-IR correlated with monocyte Fas expression at baseline in the bariatric surgery group (supplementary Fig 3) and changes in HOMA-IR 6 months after bariatric surgery significantly correlated with changes in monocyte Fas mRNA expression, even after adjustment for changes in BMI (r = 0.14, adjusted p = 0.042). Jointly, these cross-sectional and longitudinal associations between monocyte-expressed Fas and insulin resistance inspired us to hypothesize that monocyte Fas plays a causal role in obesity-associated skeletal muscle insulin resistance. To test this hypothesis, we generated myeloid-specific Fas-knockout mice.
Figure 1.
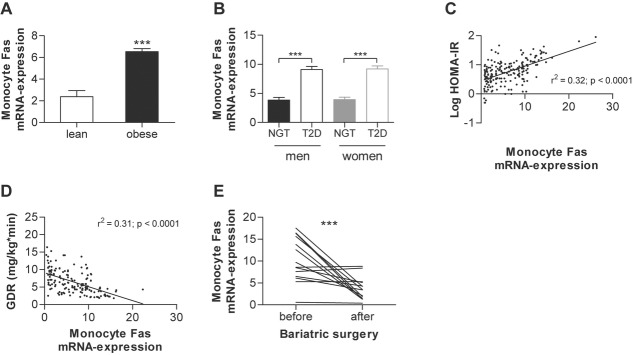
Fas expression in circulating monocytes correlates negatively with insulin sensitivity in obese patients
A Monocytes were isolated from whole human blood samples of lean and obese subjects and Fas mRNA expression was measured normalized to HPRT. n = 16–247. **p = 0.005 (Student's t-test). Error bars represent SEM.
B Monocytes were isolated from whole human blood samples and Fas mRNA expression was measured normalized to HPRT. ***p < 0.0001 (Student's t-test). NGT: normal glucose tolerance (men n = 61; women n = 70); T2D: type 2 diabetes (men n = 57; women n = 58). Error bars represent SEM.
C,D Monocyte Fas mRNA expression was determined as described above and correlated to HOMA-IR (C) and glucose disposal rate (GDR) (D). n = 200 (C) and n = 163 (D). Error bars represent SEM.
E Monocyte Fas mRNA expression was determined in obese patients before and 6 months after bariatric surgery (gastric sleeve resection; n = 14), ***p = 0.0002 (Student's t-test).
Table 1.
Basic clinical characteristics of patients
| Cross-sectional study | Bariatric surgery intervention | |||
|---|---|---|---|---|
| NGT | T2D | Baseline | 6 Months post | |
| Age | 41.3 ± 14.6 | 51.9 ± 13 | 41.9 ± 13.3 | |
| Gender (M/F) | 61/70 | 57/58 | 4/10 | |
| BMI (kg/m2) | 40.2 ± 11.7 | 47.7 ± 10.8 | 54.1 ± 8.1 | 36.3 ± 7.3b |
| Body fat (%) | 37.2 ± 11.9 | 42.1 ± 10.2 | 56.1 ± 8.3 | 38.5 ± 7.2b |
| HbA1c (%) | 5.7 ± 0.3 | 7.1 ± 1.0a | 6.6 ± 0.9 | 5.8 ± 0.35b |
| FPG (mmol/L) | 5.5 ± 0.6 | 7.2 ± 1.5a | 6.1 ± 0.6 | 5.7 ± 0.3b |
| FPI (pmol/L) | 200 ± 246 | 266 ± 233 | 382 ± 164 | 183 ± 72b |
| HOMA-IR | 7.0 ± 0.9 | 12.3 ± 2.2a | 14.9 ± 0.6 | 6.7 ± 0.1 |
| Triglycerides (mmol/L) | 2.0 ± 1.1 | 2.48 ± 1.3a | 2.3 ± 0.5 | 1.75 ± 0.3b |
| HDL-cholesterol (mmol/L) | 1.24 ± 0.3 | 1.15 ± 0.3 | 0.91 ± 0.18 | 1.23 ± 0.2b |
| hsCrP (mg/L) | 0.85 ± 1.2 | 1.1 ± 0.9 | 4.36 ± 0.7 | 2.76 ± 0.7b |
p < 0.05 for comparisons between NGT and T2D.
p < 0.05 for comparisons between 6 months post bariatric surgery and baseline.
Myeloid cell-specific Fas deletion protects from HFD-induced muscle insulin resistance
In wild-type mice, HFD-induced obesity was associated with elevated Fas levels in circulating monocytes as determined by flow cytometric analysis (Fig 2A and supplementary Fig 4). In contrast, HFD did neither increase Fas levels in B- and T-lymphocytes nor in neutrophils (supplementary Fig 5). In order to further assess a role for myeloid-expressed Fas in the development of obesity-associated insulin resistance, myeloid-specific Fas-knockout mice (Fasf/f, LysM-Cre+/−; FasΔmye) were generated using the cre-lox system (Clausen et al, 1999; Olefsky & Glass, 2010). LysM-Cre mice allow specific and highly efficient deletion of loxP-flanked target genes in myeloid cells, i.e. macrophages, monocytes, neutrophil granulocytes and partially dendritic cells (Clausen et al, 1999). As controls, littermate mice with floxed Fas but absent Cre-recombinase (Cre) expression were used (Fasf/f, LysM-Cre−/−; FasF/F). Flow cytometric analysis revealed diminished Fas protein levels in circulating monocytes and neutrophils of FasΔmye mice (Fig 2B and C). However, no reduction was observed in non-myeloid B- and T-lymphocytes (supplementary Fig 6A). Western blot analysis confirmed reduced Fas protein content in isolated macrophages of FasΔmye mice whereas it was unchanged in white adipose tissue, liver and other tissues (supplementary Fig 6B). To investigate the functional significance of myeloid-specific Fas-deletion, FasΔmye mice were fed a standard chow or HFD for 6 weeks. Total body weight gain was similar in FasΔmye and their Cre-negative littermates under both diets (Fig 2D), and no significant differences were observed in either fat pad weights or adipocyte size distribution (supplementary Fig 7A–C). Moreover, FasL expression in myeloid cells was similar between both genotypes upon HFD (supplementary Fig 8). Strikingly, whereas 6 weeks of HFD impaired glucose and insulin tolerance tests in FasF/F compared to chow-fed mice, FasΔmye mice showed no deterioration in glucose metabolism (Fig 2E and F). In addition, fasting blood glucose levels were significantly lower in HFD-fed FasΔmye compared to FasF/F littermates, whereas insulin, free fatty acid (FFA) and triglyceride (TG) levels as well as circulating adiponectin and leptin levels did not differ significantly between the two genotypes (Table 2). The protective effect against HFD-induced glucose and insulin intolerance by myeloid cell-specific Fas deletion could not be attributed to differences in food intake, locomotion or fuel utilization (respiratory quotient, RQ; supplementary Fig 9A–C).
Figure 2.

FasΔmye mice are protected from HFD-induced glucose intolerance.
A Flow cytometric analysis of peripheral blood leukocytes of chow- and HFD-fed mice. Monocytes (GR1low/im MAC1+) were stained with respective antibodies. Bar graphs show mean fluorescence intensity (MFI) of Fas (CD95) of live monocytes. n = 6–8, **p = 0.005 (Student's t-test). Error bars represent SEM.
B,C Flow cytometric analysis of circulating monocytes (B) and neutrophils (C) of Fas-deficient, FasF/F and FasΔmye mice. Cells were stained with respective antibodies and Fas fluorescence was measured.
D Body weight change during 6 weeks of chow- or HFD-fed in FasF/F and FasΔmye mice. n = 7–20. **p = 0.002 (FasF/F chow vs. HFD); **p = 0.001 (FasΔmye chow vs. HFD; Student's t-test). Error bars represent SEM.
E Intra-peritoneal glucose-tolerance test in chow- and HFD-fed FasF/F and FasΔmye mice at 12 weeks of age. n = 6–10. *p = 0.049 and 0.035 after 15 and 60 min, respectively; **p = 0.008 (ANOVA). All error bars represent SEM.
F Intra-peritoneal insulin-tolerance test in chow- and HFD-fed FasF/F and FasΔmye mice at 12 weeks of age. n = 6–10. *p = 0.034 (15 min), *p = 0.012 (30 min; ANOVA). All error bars represent SEM.
Table 2.
Phenotypic characteristics of HFD-fed FasF/F and FasΔmye mice
| FasF/F | FasΔmye | |
|---|---|---|
| Body weight (g) | 28.8 ± 0.8 | 28.2 ± 0.7 |
| Blood glucose (mmol/L) | 9.5 ± 0.5 | 8.3 ± 0.3* |
| Insulin (pmol/L) | 146.6 ± 19.9 | 120.5 ± 31.4 |
| FFA (mmol/L) | 0.77 ± 0.09 | 0.77 ± 0.08 |
| TG (mg/dl) | 117.7 ± 29.7 | 106.9 ± 13.9 |
| Adiponectin (μg/ml) | 41.4 ± 6.1 | 31.1 ± 4.6 |
| Leptin (pg/ml) | 48.1 ± 11.5 | 44.7 ± 9.8 |
| IL-6 (pg/ml) | 4.1 ± 0.7 | 2.6 ± 1.3 |
| MCP-1 (pg/ml) | 6.8 ± 0.8 | 6.4 ± 0.5 |
| IL-10 (pg/ml) | n.d. | n.d. |
| LPS (EU/ml) | 8.6 ± 1.1 | 8.3 ± 1.2 |
n.d. = not detectable.
Mice were fasted for 8 h. Results are the means ± SEM of 5–15 mice.
p = 0.049 (Student's t-test).
To better elucidate the metabolic-endocrine phenotype of HFD-fed FasΔmye mice, hyperinsulinaemic-euglycaemic clamp studies were performed. A significantly increased glucose infusion rate in HFD-fed FasΔmye compared to FasF/F mice was noted, consistent with improved whole-body insulin sensitivity (see Fig 3A and supplementary Fig 10A–C for detailed time courses). Importantly, insulin-stimulated glucose disposal rate was higher in FasΔmye compared to FasF/F littermates suggesting improved skeletal muscle insulin sensitivity (Saberi et al, 2009b; Fig 3B). Remarkably, such protective effect against insulin resistance seemed to be specific to skeletal muscle since insulin-mediated inhibition of endogenous (mainly reflecting hepatic) glucose production was not different between FasF/F and FasΔmye mice (Fig 3C) as was insulin-mediated decrease of circulating FFA levels (supplementary Fig 11). Consistent with clamp studies, insulin-stimulated glucose uptake into isolated soleus muscle was significantly increased in HFD-fed FasΔmye compared to FasF/F littermates (Fig 3D), corresponding to increased insulin-induced phosphorylation of Akt and AS160 in skeletal muscle of myeloid-specific Fas knockout mice (Fig 3E and F). In contrast, no significant differences in insulin-stimulated Akt phosphorylation were observed in white adipose tissue or liver of high fat-fed FasΔmye and FasF/F mice (supplementary Fig 12A and B). Moreover, mRNA expression of cytokines and macrophage markers in adipose tissue and liver were comparable in both groups suggesting that myeloid cell-expressed Fas does not have a major impact on adipose tissue and/or liver inflammation (supplementary Fig 12C and D). In addition, flow cytometric analysis revealed similar immune cell infiltration and macrophage polarization in adipose tissue and livers of HFD-fed control and myeloid-specific Fas knockout mice (supplementary Fig 12E–G).
Figure 3.

FasΔmye mice are protected from HFD-induced muscle insulin resistance.
A–C Glucose infusion rate (GIR), insulin-stimulated glucose disposal rate (IS GDR) and% suppression of endogenous glucose production (EGP) during hyperinsulinaemic-euglycaemic clamps, n = 4–5, *p = 0.026 (A) and *p = 0.018 (B; Student's t-test). Error bars represent SEM.
D Insulin-stimulated glucose uptake into isolated soleus muscle of HFD-fed FasF/F and FasΔmye mice relative to basal uptake. n = 4–6, *p = 0.039 (Student's t-test). Error bars represent SEM.
E,F Representative Western blots of total muscle lysates of FasF/F and FasΔmye mice. Graphs show results of 6–10 (E) and 7–8 (F) mice. *p = 0.036 (E) and *p = 0.031 (F) (Student's t-test). Error bars represent SEM.
G mRNA expression of respective genes in skeletal muscle of chow (white bars) and HFD-fed (black bars) mice. n = 4. **p = 0.002 (Student'st-test). Error bars represent SEM.
To further characterize skeletal muscle from FasΔmye mice, mRNA expression of cytokines as well as markers of β-oxidation and lipogenesis were measured, and were found to be expressed at similar levels in HFD-fed knockout and control mice (supplementary Fig 13). Importantly, there was neither a difference in inflammatory cell marker expression nor in immune cell infiltration and macrophage polarization in skeletal muscle between FasF/F and FasΔmye littermates as determined by real-time RT-PCR and flow cytometry (supplementary Fig 13A and 13B). Moreover, HFD did not induce Fas expression in skeletal muscle of C57BL6/J mice (Fig 3G) and Fas protein levels could neither be detected in skeletal muscle of HFD-fed FasF/F nor FasΔmye mice, confirming previous findings (Wueest et al, 2010b). Hence, improved skeletal muscle insulin sensitivity in myeloid-specific Fas knockout mice is likely driven by an endocrine process, rather than a paracrine effect within skeletal muscle itself.
To further confirm the causal role of myeloid Fas in obesity-associated skeletal muscle insulin resistance, we generated mice deficient of Fas in all haematopoietic-derived cells using adoptive bone marrow (BM) transfer. Wild-type C57BL6/J mice receiving a BM transplant from total body Fas-deficient (BM Fas-def) mice showed a marked reduction in Fas protein content in myeloid cells compared to BM WT mice (supplementary Fig 14). Confirming our results from myeloid-specific Fas knockout mice generated with the cre-lox technique, BM Fas-def chimeras were protected from HFD-induced skeletal muscle insulin resistance (supplementary Fig 15). Phenotypic characteristics of these mice are reported in supplementary Table S1.
Fas depletion in haematopoietic cells of ob/ob mice improves skeletal muscle insulin sensitivity
To further unravel a potential role of myeloid-expressed Fas in modulating skeletal muscle insulin resistance and to rule out any specific effects of the HFD, Fas was depleted in myeloid/haematopoietic cells of the leptin-deficient (ob/ob) mouse using adoptive BM transfer. Eight weeks after transplantation, glucose tolerance was improved in chimeric Fas-def ob/ob mice (Fig 4A). Moreover, glucose infusion rate (GIR) during hyperinsulinaemic-euglycaemic clamp studies was higher in Fas-def ob/ob compared to WT ob/ob mice (see Fig 4B for steady-state glucose infusion rates and supplementary Fig 16 for detailed time courses). Importantly, observed differences in GIR between the two groups were not due to differences in hepatic insulin sensitivity (Fig 4C), but rather to significantly improved skeletal muscle insulin sensitivity in Fas-def ob/ob mice (Fig 4D).
Figure 4.
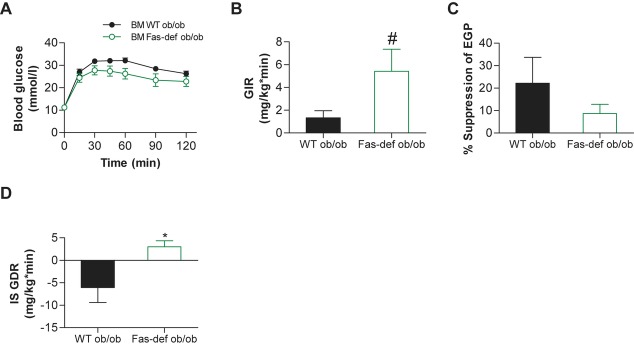
ob/ob Bone marrow chimeras with Fas-depleted myeloid cells are protected from skeletal muscle insulin resistance.
A–D (A) Intra-peritoneal glucose-tolerance test and hyperinsulinaemic-euglycaemic clamp studies (B–D) with glucose infusion rate (GIR), insulin-inhibited endogenous glucose production (EGP) and insulin-stimulated glucose disposal rate (IS GDR) were performed in ob/ob BM WT and ob/ob BM Fas-def mice. n = 5–6, #p = 0.09 (Student's t-test), *p = 0.025 (Student's t-test). All error bars represent SEM.
Jointly, these results suggest that depletion of Fas in myeloid/haematopoietic cells as demonstrated either by lineage-specific elimination or by total BM transplantation protects mice from obesity-induced skeletal muscle insulin resistance, without affecting hepatic or adipose tissue insulin sensitivity or HFD-induced weight gain.
Fas mediates myeloid-muscle cell communication leading to myocellular insulin resistance
Lipopolysaccharides (LPS) injections were previously shown to induce insulin resistance in mice (Arkan et al, 2005; Ling et al, 1994). In such treated animals, inflammation is mimicked in the absence of chronic caloric surplus and tissue infiltration with inflammatory cells. Moreover, the relevance of such approach is further strengthened by the finding that plasma LPS levels are elevated in HFD-fed mice due to increased absorption of LPS from the gut and, thus, LPS was postulated to initiate obesity and insulin resistance (Cani et al, 2007; Ley et al, 2006). In order to explore the notion whether myeloid cell-specific Fas deletion protects against LPS-induced insulin resistance, LPS was injected intraperitoneally (1 mg/kg body weight). As depicted in Fig 5A, glucose tolerance was impaired in FasF/F mice, whereas FasΔmye littermates were partly protected from LPS-induced deterioration of glucose metabolism. Of note, insulin levels after LPS injection were similar in both groups (FasF/F 131.6 ± 22.6 pmol/L vs. FasΔmye 138.0 ± 18.9 pmol/L).
Figure 5.

Fas mediates myeloid-muscle cell communication leading to myocellular insulin resistance.
A Intra-peritoneal glucose-tolerance test in FasF/F and FasΔmye mice. LPS (1 mg/kg BW) was injected 45 min prior to glucose tolerance test. n = 7, *p = 0.022, **p = 0.005, ***p = 0.0007 (Student's t-test). Error bars represent SEM.
B,C Leukocyte-mRNA expression of TNFα (n = 6–7; B) and circulating plasma TNFα levels (n = 8–13; C) of FasF/F (black bars) and FasΔmye(blue bars) mice 60 min upon intraperitoneal injection of 1 mg/kg BW LPS. *p = 0.049 (Student's t-test). Error bars represent SEM.
D Representative Western blot and quantitative analysis of total lysates of RAW cells treated with scrambled (scr, black bars) or targeted (white bars) Fas siRNA for 48 h and subsequently stimulated with or without LPS (100 ng/ml for 6 h). n = 4–5, *p = 0.01 (Mann–Whitney test, scr Co vs. scr LPS) and #p = 0.07 Student's t-test, scr LPS vs. target LPS). Error bars represent SEM.
E RAW cells were treated with scrambled (scr, black bars) or target (white bars) Fas siRNA for 48 h. Thereafter, cells were stimulated with or without 100 ng/ml LPS for 6 h and cytokine release was measured. n = 6. *p = 0.047 (scr Co vs. scr LPS) and *p = 0.042 (scr LPS vs. target LPS; Student's t-test). Error bars represent SEM.
F L6 myotubes were incubated overnight with conditioned media from RAW cells treated as mentioned above and insulin-stimulated glucose uptake was measured. n = 4–7, **p = 0.002 (scr Co vs. scr LPS; Student's t-test). Error bars represent SEM.
G L6 myotubes were incubated overnight with conditioned media from LPS-stimulated RAW cells in the presence of IgG control (open bar) or nTNFα (black bar) antibody and insulin-stimulated glucose uptake was measured. Results are expressed relative to respective unstimulated values. n = 5, *p = 0.041. Error bars represent SEM.
H Mature 3T3-L1 adipocytes were incubated overnight with conditioned media from RAW cells treated as mentioned above and insulin-stimulated glucose uptake was measured. n = 4, **p = 0.016 (scr) and **p = 0.04 (target; Student's t-test). Error bars represent SEM.
LPS was previously demonstrated to increase circulating TNFα and IL-6 levels in mice (McIlwain et al, 2012). Moreover, interruption of Fas–FasL signalling suppressed cytokine expression induced by LPS in mouse macrophages (Ma et al, 2004). As shown in Fig 5B, LPS-stimulated mRNA expression of TNFα but not IL-6 (data not depicted: 1.0 ± 0.28 vs. 0.74 ± 0.28; p = 0.51) was significantly reduced in circulating immune cells isolated from FasΔmye mice. Moreover, LPS-induced circulating TNFα levels were significantly lower in FasΔmye compared to FasF/F mice (Fig 5C). Similar to findings presented in mice, LPS increased Fas expression in RAW 264.7 cells (a murine myeloid cell line), an effect that was greatly blunted after siRNA-mediated depletion of Fas expression (Fig 5D). Concomitantly, Fas down-regulation in RAW cells significantly decreased LPS-induced release of TNFα into culture medium (Fig 5E), suggesting a role for Fas in LPS-induced TNFα secretion. In contrast to Fas, its ligand (FasL) is not induced in RAW cells after LPS stimulation (supplementary Fig 17).
To investigate a potential role of Fas in mediating myeloid-muscle inter-cellular communication, the effect of conditioned medium harvested from RAW cells was tested on insulin signalling and metabolism in L6 myotubes, mature 3T3-L1 adipocytes or HepG2 hepatocytes. Conditioned medium from RAW cells over-expressing Fas (in response to LPS stimulation) decreased insulin-stimulated glucose uptake into L6 myotubes (Fig 5F), potentially recapitulating the link between obesity-induced monocyte Fas over-expression and muscular insulin resistance (Fig 1). Fas' causal role in the process was supported by demonstrating a reversal of this effect when Fas-depleted RAW cells were used (Fig 5F). To unravel a possible role for reduced LPS-stimulated TNFα release from Fas-depleted RAW cells, insulin-stimulated glucose uptake into L6 myotubes was performed in the presence of neutralizing TNFα antibody. The inhibitory effect of LPS-treated RAW cell media on insulin-stimulated glucose uptake was blunted after TNFα neutralization (Fig 5G) suggesting a TNFα-dependent effect of conditioned media on insulin resistance. Importantly, Fas-depletion in myeloid cells had no protective effect on conditioned media-induced insulin resistance in mature 3T3-L1 adipocytes (Fig 5H) or in HepG2 hepatocytes (supplementary Fig 18).
To further unravel a causative role for TNFα in mediating obesity-induced myeloid Fas-dependent muscle insulin resistance, its expression and plasma levels were analysed. As shown in Fig 6A, TNFα expression was significantly reduced in circulating immune cells isolated from HFD-fed FasΔmye compared to FasF/F mice. Moreover, circulating TNFα concentrations were significantly reduced in HFD-fed FasΔmye mice (Fig 6B), while plasma IL-6 levels where not different. Of note, plasma LPS levels were similar in both genotypes (Table 2). Likewise, circulating TNFα levels were significantly lower in HFD-fed BM Fas-def mice (supplementary Table S1) and reduced in Fas-def ob/ob mice (4.5 ± 0.5 pg/ml in Fas-def ob/ob vs. 5.6 ± 0.9 pg/ml in WT ob/ob). Moreover, treatment of HFD-fed FasF/F and FasΔmye littermates with the TNFα blocking monoclonal antibody infliximab (Araujo et al, 2007), which recognizes both human as well as mouse TNFα epitopes (Qiu et al, 2011) abolished HFD-induced differences in glucose tolerance between both groups (Fig 6C) supporting the notion that circulating TNFα, induced by monocytic Fas, contributes to HFD-induced glucose intolerance.
Figure 6.
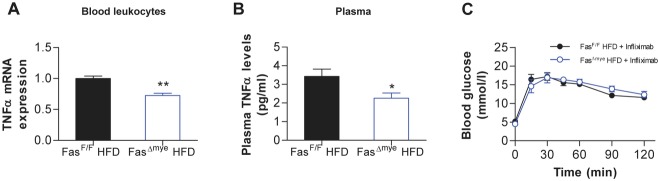
Reduced circulating TNFα levels in FasΔmye mice.
A,B Leukocyte-mRNA expression of TNFα (n = 3–5; A) and circulating plasma TNFα levels (n = 12–15; B) of HFD-fed FasF/F (black bars) and FasΔmye (blue bars) mice. *p = 0.033, **p = 0.003 (Student's t-test). Error bars represent SEM.
C Intra-peritoneal glucose tolerance test in HFD-fed FasF/F and FasΔmyemice pre-treated with the TNFα neutralizing antibody infliximab. n = 4. All error bars represent SEM.
Human monocyte Fas expression correlates positively with serum LPS and TNFα
In order to evaluate a potential involvement of TNFα in mediating the effect of monocyte-specific Fas on skeletal muscle insulin resistance, we determined circulating TNFα and LPS levels in the obese human cohort. Monocyte Fas mRNA positively correlated with circulating LPS (Fig 7A) and TNFα levels (Fig 7B). Multivariate linear regression models revealed that the correlations of monocytic Fas expression with LPS and TNFα serum concentrations are independent of age, gender and BMI (supplementary Table S2). In addition, weight loss in obese patients 6 months after bariatric surgery was associated with reduced circulating LPS and TNFα levels (Fig 7C and D) and BMI-adjusted changes in monocyte Fas expression significantly correlate with changes in circulating LPS (r = 0.14, p = 0.045) and TNFα (r = 0.15, p = 0.04). Jointly, these data support a role for monocyte-expressed Fas in the development of obesity-associated (muscle) insulin resistance in humans potentially via increased release of TNFα into circulation.
Figure 7.
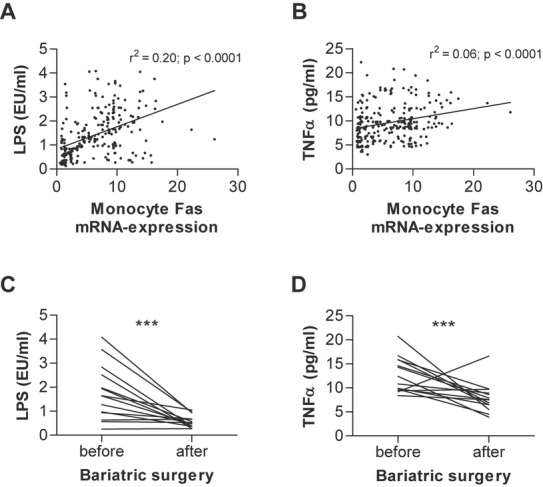
Human monocyte Fas expression positively correlates with serum LPS and TNFα levels.
A,B Monocyte Fas mRNA expression was determined and correlated to circulating LPS and TNFα levels. n = 246.
C,D Plasma LPS and TNFα levels were determined in obese patients before and 6 months after bariatric surgery (gastric sleeve resection; n = 14), ***p = 0.0007 (C) and ***p = 0.0009 (D) (Student's t-test).
Discussion
Unraveling inter-organ and inter-cell type specific crosstalk pathways underlying obesity-associated insulin resistance not only enhances our understanding of this complex disease process, but can yield new pharmaceutical interventions. The “inflammatory basis of obesity-associated morbidity” encompasses a particular challenge, since it involves activation of inflammatory cascades in multiple haematopoietic and non-haematopoietic cell types, and includes systemic and local markers of low-grade and chronic inflammatory activation that are all tightly inter-connected. The present study revealed a pathway in which myeloid cell-expressed Fas regulates systemic levels of TNFα. The latter in turn determines whole-body insulin sensitivity, particularly by modulating skeletal muscle insulin responsiveness. The major findings of this study supporting this proposition are: (i) myeloid cell-specific Fas disruption in mice results in lower circulating levels of TNFα, with improved whole-body insulin sensitivity that can be largely assigned to skeletal muscle; (ii) in obese human subjects, monocyte Fas expression correlates with circulating levels of TNFα and with glucose disposal rate. Demonstrating the protective effect of myeloid Fas ablation on skeletal muscle insulin sensitivity in the context of diet-induced obesity and leptin-deficiency as well as in response to LPS suggests the possibility that myeloid Fas is not an initiator of inflammation in obesity, but rather an ‘intermediate integrator’ that may respond to inflammatory cues like increased plasma LPS levels in the obese state. Thus, the myeloid cell—muscular crosstalk unraveled here is likely merely a segment of a more intricate pathway, in which myeloid Fas may constitute a critical node.
A specific example for the regulatory role of Fas is exemplified by the response to LPS. LPS was previously found to induce inflammatory cytokines in monocytes and macrophages, and interruption of Fas–FasL signalling suppressed nuclear factor-κB activation and cytokine expression induced by LPS in primary human and mouse macrophages (Ma et al, 2004). Complementarily, Fas activation induced a rapid release of pro-inflammatory cytokines in macrophages (Wang et al, 2010) and human monocytes (Park et al, 2003), which was dependent on toll-like receptor 4 (TLR4), a major receptor of LPS. We show here that LPS-induced up-regulation of Fas protein levels in myeloid cells significantly contributes to TNFα release. In vivo, high fat dieting (possibly via increased circulating LPS levels) increases myeloid Fas protein expression, and myeloid-specific Fas knockout reduces HFD- and LPS-induced circulating TNFα but not IL-6 levels. Hence, our data suggest that myeloid-cell expressed Fas plays an important role in the regulation of circulating TNFα levels. Moreover, our data provide evidence for a similar role of monocytic Fas in humans: monocyte Fas expression in obese people correlated with circulating LPS as well as TNFα concentrations and the drop in LPS levels in patients after bariatric surgery was paralleled by reduced monocytic Fas and circulating TNFα levels as well as improved insulin sensitivity. As previously reported, the composition of gut microbiota changes after bariatric surgery (Clement, 2011; Furet et al, 2010) and, thus, the observed improvement of glucose metabolism may at least in part be due to reduced endotoxemia and consequently reduced LPS levels (Manco et al, 2010). A role for Fas in metabolic diseases in human is further supported by a recent study revealing an association of Fas and FasL gene promoter polymorphisms with type 2 diabetes (Nolsoe et al, 2006).
TNFα has been among the first inflammatory mediators to be implicated in the induction of insulin resistance in obesity (Hotamisligil et al,1993). While TNFα infusion in healthy humans had no effect on endogenous glucose production, it induced skeletal muscle insulin resistance via inhibition of Akt substrate 160 phosphorylation (Plomgaard et al, 2005). Similarly, we found here that the decrease in circulating TNFα levels in mice with myeloid-specific deletion of Fas was associated with improved skeletal muscle but not liver insulin sensitivity. Moreover, neutralization of TNFα blunted HFD-induced differences in glucose tolerance between myeloid-specific Fas knockout and control mice, further supporting a causative role for TNFα in the observed phenotype. Furthermore, monocytic Fas expression in obese human individuals correlated positively with circulating TNFα concentration suggesting that the latter might be one of the downstream effectors of Fas up-regulation in myeloid cells in obesity-induced insulin resistance in human individuals. Of note, TNFα neutralization was reported to improve insulin resistance in diabetic patients with rheumatoid arthritis or Crohn's disease who were also receiving anti-TNFα treatment (infliximab) for their autoimmune disease (Gupta-Ganguli et al, 2011). Since we found no significant difference in immune cell infiltration and local cytokine expression in fat, liver and skeletal muscle between HFD-fed FasF/F and FasΔmye littermates, Fas expression in monocytes does not seem to impact on monocyte migration into peripheral tissue, supporting the idea that monocyte recruitment is mainly guided by recipient tissue rather than properties of circulating monocytes (Oh et al, 2012). Moreover, our findings would also suggest that myeloid cells rather than adipose tissue are the main source for circulating TNFα. Of note, the chosen period of high fat feeding (6 weeks) in our study is rather short. We cannot exclude that longer exposure to HFD may have led to a higher degree of adipose tissue inflammation and hepatic steatosis (Strissel et al, 2007), and, hence, myeloid-specific Fas deletion may have also influenced adipose tissue inflammation and/or hepatic insulin resistance.
The effect of LPS on cytokine expression is mainly mediated via TLR4, a pattern-recognition receptor known to regulate release of pro-inflammatory cytokines such as TNFα and IL-6 from monocytes/macrophages (Nguyen et al, 2007; Shi et al, 2006). As mentioned above, Fas was previously shown to modulate/enhance TLR4-mediated cytokine production (Ma et al, 2004). Hence, the beneficial effect of myeloid-specific Fas depletion on glucose tolerance may be merely due to reduced TLR4-signalling. However, myeloid/haematopoietic cell-specific TLR4 knockout mice as generated by adoptive BM transfer were not protected from HFD-induced skeletal muscle insulin resistance, whereas hepatic insulin sensitivity was significantly improved (supplementary Fig 19). Our data are in agreement with a previous study revealing blunted HFD-induced adipose and liver inflammation and ameliorated obesity-induced adipose and liver insulin resistance but no improvement in skeletal muscle insulin sensitivity in mice with haematopoietic cell-specific deletion of TLR4 (Saberi et al, 2009b). However, in contrast to this study (Saberi et al, 2009b), we found no difference in GIR between chimeras (supplementary Fig 19), which might be explained by different periods of HFD (6 weeks vs. 16 weeks). Of note, TNFα levels in HFD-fed haematopoietic cell-specific TLR4KO mice were comparable to wild-type chimeras (2.70 ± 0.29 pg/ml vs. 2.49 ± 0.3.5 pg/ml; p = 0.63).
In conclusion, our results reveal an important and unique role for myeloid cell expressed Fas in mediating obesity-induced insulin resistance in skeletal muscle. Pharmaceutical inhibition of Fas or its signalling pathway in myeloid cells may emerge as a promising new avenue in the treatment of insulin resistance and therefore type 2 diabetes.
Materials and Methods
Human samples
A total of 246 Caucasian men (n = 118) and women (n = 128) were selected among ∼1500 subjects recruited in the context of a study on insulin resistance at the Department of Medicine, University of Leipzig to represent a wide range of obesity, insulin sensitivity, and glucose tolerance. The age ranged from 19 to 80 years and BMI from 17.1 to 79.1 kg/m2. Subjects were subsequently divided into groups of NGT and type 2 diabetes (T2D) according to ADA-criteria (Anonymous, 2011). All study protocols have been approved by the ethics committee of the University of Leipzig. All participants gave written informed consent before taking part in the study.
Bariatric surgery study
Fourteen Caucasian obese volunteers (10 females, 4 males) participated in a prospective weight loss study before and 6 months after gastric sleeve resection. The baseline BMI was 54 ± 8 kg/m2 and the BMI 6 months after bariatric surgery was 36.3 ± 7.3 kg/m2. Individuals fulfilled the following inclusion criteria: (i) absence of any acute or chronic inflammatory disease as determined by a leukocyte count >7000 Gpt/L, C-reactive protein (CRP) >5.0 mg/dl or clinical signs of infection, (ii) undetectable antibodies against glutamic acid decarboxylase (GAD), (iii) no clinical evidence of either cardiovascular or peripheral artery disease, (iv) No thyroid dysfunction, (v) no alcohol or drug abuse, (vi) no pregnancy. BMI was calculated as weight divided by squared height. Hip circumference was measured over the buttocks; waist circumference was measured at the midpoint between the lower ribs and iliac crest.
Fas/FasL mRNA expression studies
Human monocytes were obtained from heparinized blood. Monocytes were separated using superparamagnetic polystyrene beads coated with a primary monoclonal antibody specific for the CD14 membrane antigen expressed on human monocytes (Invitrogen, Groningen, Germany) and resuspended in Hanks balanced solution containing (in mmol/L) NaCl 136, KCl 5.40, CaCl2 1, KH2PO4 0.44, Na2HPO4 0.34, HEPES 10, pH 7.4. Total RNA was isolated from monocytes using the RNeasy mini kit including RNase-free DNase set (Qiagen, Hilden, Germany). Using the transcriptor first-strand cDNA synthesis kit (Roche Diagnostics, Mannheim, Germany), cDNA was synthesized from 2 μg of total RNA using oligo dT (12–18). Human Fas and FasL mRNA expression, determined by a premixed assay on demand (PE Biosystems, Darmstadt, Germany) was calculated relative to the mRNA expression of HPRT (PE Biosystems). The specificity of the PCR was verified by subjecting the amplification products to agarose gel electrophoresis.
Analysis of LPS, IL-6 and TNFα serum concentrations in human cohorts
All baseline blood samples were collected between 8 and 10 am after an overnight fast. Serum IL-6 and TNFα concentrations were measured by highly sensitive ELISAs for IL-6 (Quantikine IL-6, R&D Systems, Oxford, UK) and TNFα (Alpco Diagnostics, Salem, NH, USA). Serum LPS concentration was measured using a LAL Chromogenic Endpoint Assay (Hycult Biotech Inc., PB Uden, The Netherlands).
Animals
Total body Fas-deficient (Fas-def) mice on C57BL/6J inbred strain background (B6.MRLFaslpr) were obtained from The Jackson Laboratory. C57BL/6J mice with exon IX of Fas flanked with LoxP sites were produced as described (Stranges et al, 2007) and crossed to LysozymeM (LysM)-Cre mice (B6,129-Lys<tm1(Cre)Ifo>) to generate myeloid-specific deletions (FasΔmye). All mice were housed in a specific pathogen-free environment on a 12-h light-dark cycle and fed ad libitum with regular chow diet (Provimi Kliba) or HFD (58 kcal% fat w/sucrose Surwit Diet, D12331, Research Diets). All protocols conformed to the Swiss animal protection laws and were approved by the Cantonal Veterinary Office in Zurich, Switzerland.
Genotyping of animals
All mice were genotyped by PCR with primers amplifying the Cre transgene (generating 350 bp wild-type and 700 bp Cre allele products) and Fas (generating 319 bp wild-type and 399 bp ‘floxed’ allele products).
Bone marrow transfer
BM chimeras were generated by either transplanting 2 × 106 B6.MRLFaslpr/J CD45.2 (FAS-KO), Tlr4tm1Aki (CD45.2) or C57BL/6J CD45.2 (control) total BM cells into lethally irradiated (2 × 450 cGy) 6-week-old B6.SJL CD45.1 or 6-week-old B6.V-Lepob/OlaHsd recipient mice. Recipient mice were maintained and bled at week 6 to check the chimerism in peripheral blood and a > 90% donor chimerism was confirmed in peripheral blood of all recipient animals.
Metabolic cage analysis
Locomotion, food and water intake, O2 consumption and CO2 production were determined for single housed mice during a 24-h period in a metabolic and behavioural monitoring system (PhenoMaster, TSE Systems, Bad Homburg, Germany).
Intra-peritoneal glucose and insulin tolerance tests
For the intraperitoneal glucose tolerance test (ipGTT) mice were either fasted for 5 h (ob/ob mice) or overnight (C57BL/6J), for the intraperitoneal insulin tolerance tests (ipITT) mice were fasted for 3 h. Either glucose (0.25 g/kg body weight for ob/ob mice; 2 g/kg body weight for C57BL/6J) or human recombinant insulin (0.75 U/kg body weight) were injected intraperitoneally (Konrad et al, 2007).
Glucose clamp studies
Glucose clamp studies were performed as described (Rytka et al, 2011). Clamps were performed in freely moving mice. Glucose infusion rate was calculated once glucose infusion reached a more or less constant rate with blood glucose levels at 5 mmol/L (80–90 min after the start of insulin infusion). Thereafter, blood glucose was kept constant at 5 mmol/L for 20 min and glucose infusion rate was calculated. The glucose disposal rate was calculated by dividing the rate of [3-3H]glucose infusion by the plasma [3-3H]glucose specific activity (Fisher & Kahn, 2003; Kim et al, 2000). Endogenous glucose production during the clamp was calculated by subtracting the glucose infusion rate from the glucose disposal rate (Fisher & Kahn, 2003; Kim et al, 2000). Insulin-stimulated glucose disposal rate was calculated by subtracting basal endogenous glucose production (equal to basal glucose disposal rate) from glucose disposal rate during the clamp (Saberi et al, 2009a).
Glucose incorporation into isolated soleus muscle
Intact soleus muscle was isolated and thereafter incubated with or without 2 mU of insulin per ml. 2-Deoxyglucose uptake was then measured for 20 min as described previously (Rudich et al, 2003).
Determination of plasma insulin, adipokines, free fatty acid and triglyceride levels
Plasma insulin and FFA levels were determined as described (Konrad et al, 2007). Plasma leptin and cytokine levels were determined with mouse procarta cytokine assay kit (Affymetrix). Plasma adiponectin levels were measured with an ELISA kit (Axxora). TG concentrations were determined using a colorimetric assay (Sigma).
TNFα neutralization
Mice were treated with infliximab (Remicade®, MSD) for 8 days by daily injections (i.p.) of 50 μg infliximab dissolved in 100 μl saline.
Stromal vascular fraction (SVF), muscle and liver cell isolation for flow cytometry
After digestion of adipose tissue (Rudich et al, 2003), isolated cells were centrifuged at 4°C (500 g) for 5 min. SVF (pellet) was kept on ice in KRP 0.1% BSA. Centrifugation step was repeated with infra- and supernatant for three times and obtained SVFs were pooled and dissolved in PBS 0.1% BSA. For isolation of muscle cells, skeletal muscle was minced in PBS and kept on ice. After centrifugation, 5 ml collagenase solution (0.5 ml 10 × HBSS, 4.5 ml H2O, 75 mg BSA, 10 mg collagenase II (Life Technologies)) was added to the pellet and shaken in a water bath at 37°C for 20 min. Thereafter, cells were centrifuged (80 g) and supernatant was poured into 15 ml ice-cold media (DMEM low glucose, 10% FBS). Digestion of the pellet was repeated twice. Subsequently, cells were filtered through a cell restrainer (100 μm), centrifuged (350 g), resuspended in erythrocyte lysis buffer and filled up with ice-cold PBS to 25 ml. After filtering through a 40 μm cell restrainer, cell pellet was dissolved in PBS 0.1% BSA. For liver cell isolation, livers were mashed between glass slides and cells were suspended in FACS buffer. Cell suspension was layered on Ficoll and centrifuged (400 g) for 30 min at 18°C. Cells on top of Ficoll were collected and washed in FACS buffer.
Flow cytometric analysis
For flow cytometric analysis of isolated SVF, muscle and liver cells, cells were blocked with Fc blocker (anti CD16/32 BD pharmingen) on ice for 15 min. Thereafter, cells were stained on ice for 30 min with the following antibodies: CD206-Alexa Fluor 488, CD11c-PE Cy7 (both from Biolegend), CD45.2-PE, CD8-PE-Cy5, CD4-APC (all from eBioscience), F4/80-Fluor 780 (Alexa). Cells were subsequently washed for three times and resuspended in Hoechst buffer. For flow cytometry analysis of circulating blood cells, mice were bled and red blood cells were lysed using ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). Cell debris was removed by filtering the cell suspension using a 70 μm cell strainer (BD). Peripheral leukocytes were stained using fluorochrome conjugated antibodies as follows: Pacific Blue-conjugated anti-CD45.1 (clone A20; Biolegend), FITC-conjugated anti-GR-1 (clone RB6-8C5; ebioscience.com), PE-conjugated anti-FAS (CD95; clone Jo2; BD-Pharmingen), PE-Cy5-conjugated anti-CD45.2 (clone 104; ebioscience), APC-conjugated anti-CD11c (clone M1/70; ebioscience), eFluor 780 conjugated anti-CD11b (clone M1/70; ebioscience) and Hoechst 33342 2 μg/ml to exclude dead cells. Immunophenotypical analysis was performed on a BD FACS Canto II Flow Cytometer.
Histology
Fat tissues were fixed in 4% buffered formalin and embedded in paraffin. Sections were cut and stained with haematoxylin and eosin. For each fat pad of a mouse, at least 100 adipocytes were analysed (ImageJ software; National Institutes of Health, Bethesda, MD).
RNA extraction and quantitative reverse transcription-PCR (RT-PCR)
Total RNA was extracted and reverse transcribed as described (Wueest et al, 2010b). The following primers were used: TNF-α Mm00443258_m1, IL-6 Mm00446190_m1, IL-1β Mm0043422/8_m1, cd11b Mm00434455_m1, cd11c Mm00498698_m1, cd68 Mm03047343_m1, F4/80 Mm00802529_m1, MCP-1 Mm00441242_m1, IL-1β Mm0043422/8_m1, IL-10 Mm00439614_m1, FAS Mm00662319_m1, ACC-1 Mm01304289_m1, CPT-1 Mm00550438_m1, AOX Mm00443579_m1, PGC-1α Mm01208835_m1, FasL Mm00438864_m1 (Applied Biosystems).
Western blotting
Cells or tissues were lysed and Western blots were performed as previously described (Wueest et al, 2010b). The following primary antibodies were used: anti-Fas (clone 7C10), anti-phospho-AS160 (Thr642) and anti-actin (Millipore), anti-phospho-Akt (Thr308; Cell Signaling).
Cell culture experiments
RAW 264.7 cells were grown in RPMI 1640, 1% penicillin/streptomycin (p/s), 1% glutamine and 10% FCS. At 30–50% confluence, cells were treated with siRNA (target sequences: 5′ GAACCATTATGCTGATAAA 3′, 5′ AAAGCCGAATGTCGCAGAA 3′; scrambled sequences: 5′ GACACTAAACGTGAATTAT 3′, 5′ GCAACAGGCGTCAAGAAAT 3′) in a transfection mixture containing Lipofectamin 2000 (Invitrogen) in RPMI (without FCS, without p/s) according to the manufacturer's instructions. L6 myotubes differentiation and glucose uptake were performed as described (Niu et al, 2003). For TNFα neutralization, L6 myotubes were incubated with nTNFα or IgG control antibody (R&D Systems). Glucose uptake in mature 3T3-L1 adipocytes was performed as described (Wueest et al, 2010a). HepG2 cells were cultivated in DMEM supplemented with 10% FCS.
Data analysis
Animal data
Data are presented as means ± SEM and were analysed by two-tailed Student's t-test, one sample t-test or one-way ANOVA with a Tukey correction for multiple group comparisons. p Values <0.05 were considered significant.
Human cohort data
Data are shown as means ± SEM unless stated otherwise. Before statistical analysis, non-normally distributed parameters were logarithmically transformed to approximate a normal distribution. The following statistical tests were used: paired Student's t-test, Chi-square test, and Pearson's simple correlation. Prediction models for monocyte Fas mRNA expression based on age, gender, anthropometric and clinical parameters were calculated by multivariate linear regression analysis. p Values <0.05 were considered to be statistically significant. Statistical analysis was performed using SPSS version 12.0 (Chicago, IL).
Acknowledgments
This work was supported by grants from the Swiss National Science Foundation (#310000-112275), the European Foundation for the Study of Diabetes (EFSD-Lilly; all to D.K.), the Foundation for Research at the Medical Faculty, University of Zurich (to F.I.) and the Promedica Foundation (Chur, Switzerland; to M.G.M.). We would like to greatly acknowledge Prof. Giatgen Spinas, University Hospital Zurich, for continuous support, PD Dr. Maggy Arras, University Hospital Zurich, for expert veterinary advice, Prof. Assaf Rudich, Ben-Gurion University of the Negev, Israel, for very helpful discussion, Prof. Adriano Fontana for providing LysM-Cre expressing mice, Prof. Amira Klip and Dr. Philip Bilan, Hospital for Sick Children and University of Toronto, Canada, for providing L6 myotubes, Dr. Richard Zuellig, University Hospital Zurich, for expert advice on LPS determination, Prof. Jan Krützfeldt, Ms. Katarina Turcekova and Ms. Tatiane Gorski, University Hospital Zurich, for expert advice regarding isolation of skeletal muscle cells, Mrs. Christina Murer-Pesenti, MSD Switzerland, for allocating Infliximab (Remicade®) and Dr. Henrike Sell, Deutsches Diabetes Zentrum (DDZ) Düsseldorf, Germany, for expert advice regarding cell co-culture experiments.
Author contributions
SW and DK conceived the study and wrote the paper. SW, RM, MB, FI, ASHC, MSFW, HT, LK and DK performed the experimental work. AVC provided the floxed Fas mice and gave conceptual advice. EJS and MGM gave conceptual advice. All authors contributed to discussion and reviewed/edited manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
Low grade inflammation in adipose tissue and liver has been shown to contribute to the development of obesity-associated insulin resistance and type 2 diabetes. However, it is presently unknown whether inflammatory cells play a role in the pathogenesis of skeletal muscle insulin resistance.
Results
Herein we demonstrate that expression of Fas (CD95) in circulating monocytes correlate with skeletal muscle insulin resistance in a large cohort of obese human individuals. To test a causal role of monocyte-expressed Fas in the development of skeletal muscle insulin resistance, we generated mice lacking Fas in myeloid cells. Such mice were protected from the development of obesity-induced skeletal muscle insulin resistance whereas adipose tissue inflammation and hepatic insulin resistance were unaltered. Mechanistically, we provide evidence that activation of Fas in myeloid cells mediate the release of TNFα inducing skeletal muscle insulin resistance. Accordingly, monocyte Fas expression correlated with circulating TNFα levels in humans.
Impact
Our results demonstrate an unanticipated crosstalk between myeloid cells and skeletal muscle in the development of obesity-associated insulin resistance. Pharmaceutical inhibition of Fas or its signalling pathway in myeloid cells may emerge as a promising future strategy to prevent or treat type 2 diabetes.
References
- Anonymous. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2011;34:S62–S69. doi: 10.2337/dc11-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo EP, Souza De CT, Ueno M, Cintra DE, Bertolo MB, Carvalheira JB, Saad MJ, Velloso LA. Infliximab restores glucose homeostasis in an animal model of diet-induced obesity and diabetes. Endocrinology. 2007;148:5991–5997. doi: 10.1210/en.2007-0132. [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Clement K. Bariatric surgery, adipose tissue and gut microbiota. Int J Obes (London) 2011;35:S7–S15. doi: 10.1038/ijo.2011.141. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32:S157–S163. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Faouzi S, Burckhardt BE, Hanson JC, Campe CB, Schrum LW, Rippe RA, Maher JJ. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J Biol Chem. 2001;276:49077–49082. doi: 10.1074/jbc.M109791200. [DOI] [PubMed] [Google Scholar]
- Farley SM, Purdy DE, Ryabinina OP, Schneider P, Magun BE, Iordanov MS. Fas ligand-induced proinflammatory transcriptional responses in reconstructed human epidermis. Recruitment of the epidermal growth factor receptor and activation of MAP kinases. J Biol Chem. 2008;283:919–928. doi: 10.1074/jbc.M705852200. [DOI] [PubMed] [Google Scholar]
- Fisher SJ, Kahn CR. Insulin signaling is required for insulin's direct and indirect action on hepatic glucose production. J Clin Inves. 2003;111:463–468. doi: 10.1172/JCI16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, Mariat D, Corthier G, Dore J, Henegar C, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59:3049–3057. doi: 10.2337/db10-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zhang M, Tang H, Cao X. Fas signal links innate and adaptive immunity by promoting dendritic-cell secretion of CC and CXC chemokines. Blood. 2005;106:2033–2041. doi: 10.1182/blood-2004-12-4831. [DOI] [PubMed] [Google Scholar]
- Gupta-Ganguli M, Cox K, Means B, Gerling I, Solomon SS. Does therapy with anti-TNF-alpha improve glucose tolerance and control in patients with type 2 diabetes? Diabetes Care. 2011;34:e121. doi: 10.2337/dc10-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Imamura R, Konaka K, Matsumoto N, Hasegawa M, Fukui M, Mukaida N, Kinoshita T, Suda T. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem. 2004;279:46415–46423. doi: 10.1074/jbc.M403226200. [DOI] [PubMed] [Google Scholar]
- Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, Kahn CR, Shulman GI. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105:1791–1797. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad D, Rudich A, Schoenle EJ. Improved glucose tolerance in mice receiving intraperitoneal transplantation of normal fat tissue. Diabetologia. 2007;50:833–839. doi: 10.1007/s00125-007-0596-1. [DOI] [PubMed] [Google Scholar]
- Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Ling PR, Bistrian BR, Mendez B, Istfan NW. Effects of systemic infusions of endotoxin, tumor necrosis factor, and interleukin-1 on glucose metabolism in the rat: relationship to endogenous glucose production and peripheral tissue glucose uptake. Metabolism. 1994;43:279–284. doi: 10.1016/0026-0495(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Ma Y, Liu H, Tu-Rapp H, Thiesen HJ, Ibrahim SM, Cole SM, Pope RM. Fas ligation on macrophages enhances IL-1R1-Toll-like receptor 4 signaling and promotes chronic inflammation. Nat Immunol. 2004;5:380–387. doi: 10.1038/ni1054. [DOI] [PubMed] [Google Scholar]
- Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31:817–844. doi: 10.1210/er.2009-0030. [DOI] [PubMed] [Google Scholar]
- Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Bluher M, Kolanus W, Kahn CR, Bruning JC. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010;6:e1000938. doi: 10.1371/journal.pgen.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T, Murthy A, Duncan G, Xu HC, et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science. 2012;335:229–232. doi: 10.1126/science.1214448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa K, Asano M, Horai R, Iwakura Y, Nagata S, Suda T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med. 1998;4:1287–1292. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- Niu W, Huang C, Nawaz Z, Levy M, Somwar R, Li D, Bilan PJ, Klip A. Maturation of the regulation of GLUT4 activity by p38 MAPK during L6 cell myogenesis. J Biol Chem. 2003;278:17953–17962. doi: 10.1074/jbc.M211136200. [DOI] [PubMed] [Google Scholar]
- Nolsoe RL, Hamid YH, Pociot F, Paulsen S, Andersen KM, Borch-Johnsen K, Drivsholm T, Hansen T, Pedersen O, Mandrup-Poulsen T. Association of a microsatellite in FASL to type II diabetes and of the FAS-670G>A genotype to insulin resistance. Genes Immun. 2006;7:316–321. doi: 10.1038/sj.gene.6364300. [DOI] [PubMed] [Google Scholar]
- Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased macrophage migration into adipose tissue in obese mice. Diabete. 2012;61:346–354. doi: 10.2337/db11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Park DR, Thomsen AR, Frevert CW, Pham U, Skerrett SJ, Kiener PA, Liles WC. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol. 2003;170:6209–6216. doi: 10.4049/jimmunol.170.12.6209. [DOI] [PubMed] [Google Scholar]
- Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, Owen LB, Pope RM, Tschopp J, Wajant H, et al. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–450. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes. 2005;54:2939–2945. doi: 10.2337/diabetes.54.10.2939. [DOI] [PubMed] [Google Scholar]
- Qiu W, Wu B, Wang X, Buchanan ME, Regueiro MD, Hartman DJ, Schoen RE, Yu J, Zhang L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest. 2011;121:1722–1732. doi: 10.1172/JCI42917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudich A, Konrad D, Török D, Ben-Romano R, Huang C, Niu W, Garg RR, Wijesekara N, Germinario RJ, Bilan PJ, et al. Indinavir uncovers different contributions of GLUT4 and GLUT1 towards glucose uptake in muscle and fat cells and tissues. Diabetologia. 2003;46:649–658. doi: 10.1007/s00125-003-1080-1. [DOI] [PubMed] [Google Scholar]
- Rytka JM, Wueest S, Schoenle EJ, Konrad D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes. 2011;60:56–63. doi: 10.2337/db10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi M, Bjelica D, Schenk S, Imamura T, Bandyopadhyay G, Li P, Vargeese C, Wang W, Bowman K, Zhang Y, et al. Novel liver-specific TORC2 siRNA corrects hyperglycemia in rodent models of type 2 diabetes. Am J Physiol Endocrinol Metab. 2009a;297:E1137–E1146. doi: 10.1152/ajpendo.00158.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi M, Woods NB, de LucaC, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009b;10:419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samokhvalov V, Bilan PJ, Schertzer JD, Antonescu CN, Klip A. Palmitate- and lipopolysaccharide-activated macrophages evoke contrasting insulin responses in muscle cells. Am J Physiol Endocrinol Metab. 2009;296:E37–E46. doi: 10.1152/ajpendo.90667.2008. [DOI] [PubMed] [Google Scholar]
- Schaub FJ, Liles WC, Ferri N, Sayson K, Seifert RA, Bowen-Pope DF. Fas and Fas-associated death domain protein regulate monocyte chemoattractant protein-1 expression by human smooth muscle cells through caspase- and calpain-dependent release of interleukin-1alpha. Circ Res. 2003;93:515–522. doi: 10.1161/01.RES.0000093205.42313.7C. [DOI] [PubMed] [Google Scholar]
- Schumann DM, Maedler K, Franklin I, Konrad D, Storling J, Boni-Schnetzler M, Gjinovci A, Kurrer MO, Gauthier BR, Bosco D, et al. The Fas pathway is involved in pancreatic beta cell secretory function. Proc Natl Acad Sci USA. 2007;104:2861–2866. doi: 10.1073/pnas.0611487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunit. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, II, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 2003;14:53–66. doi: 10.1016/s1359-6101(02)00072-2. [DOI] [PubMed] [Google Scholar]
- Wang F, Lu Z, Hawkes M, Yang H, Kain KC, Liles WC. Fas (CD95) induces rapid, TLR4/IRAK4-dependent release of pro-inflammatory HMGB1 from macrophages. J Inflamm (Lond) 2010;7:30. doi: 10.1186/1476-9255-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wueest S, Rapold RA, Schoenle EJ, Konrad D. Fas activation in adipocytes impairs insulin-stimulated glucose uptake by reducing Akt. FEBS Lett. 2010a;584:4187–4192. doi: 10.1016/j.febslet.2010.08.052. [DOI] [PubMed] [Google Scholar]
- Wueest S, Rapold RA, Schumann DM, Rytka JM, Schildknecht A, Nov O, Chervonsky AV, Rudich A, Schoenle EJ, Donath MY, et al. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Inves. 2010b;120:191–202. doi: 10.1172/JCI38388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.