

Abstract
mRNA alternative polyadenylation (APA) plays a critical role in post-transcriptional gene control and is highly regulated during development and disease. However, the regulatory mechanisms and functional consequences of APA remain poorly understood. Here, we show that an mRNA 3′ processing factor, Fip1, is essential for embryonic stem cell (ESC) self-renewal and somatic cell reprogramming. Fip1 promotes stem cell maintenance, in part, by activating the ESC-specific APA profiles to ensure the optimal expression of a specific set of genes, including critical self-renewal factors. Fip1 expression and the Fip1-dependent APA program change during ESC differentiation and are restored to an ESC-like state during somatic reprogramming. Mechanistically, we provide evidence that the specificity of Fip1-mediated APA regulation depends on multiple factors, including Fip1-RNA interactions and the distance between APA sites. Together, our data highlight the role for post-transcriptional control in stem cell self-renewal, provide mechanistic insight on APA regulation in development, and establish an important function for APA in cell fate specification.
Keywords: alternative polyadenylation, mRNA processing, reprogramming, stem cell
Introduction
Approximately 70% of mammalian genes produce alternatively polyadenylated mRNAs, which may encode different proteins and/or contain different 3′ untranslated regions (UTRs) (Di Giammartino et al, 2011; Shi, 2012; Elkon et al, 2013; Mueller et al, 2013; Tian & Manley, 2013). The diverse 3′ UTRs generated by alternative polyadenylation (APA) may impact the stability, translation, and/or intracellular localization of mRNAs, thereby modulating the protein output of gene expression (Di Giammartino et al, 2011; Shi, 2012; Elkon et al, 2013; Tian & Manley, 2013). Recent global studies revealed that APA is dynamically regulated in development and in response to environmental stimuli, and de-regulation of APA has been associated with a number of human diseases (Di Giammartino et al, 2011; Shi, 2012; Elkon et al, 2013; Mueller et al, 2013; Tian & Manley, 2013). Interestingly, some cell types, including stem cells and cancer cells, generally favor upstream or proximal polyadenylation sites (PASs) and produce mRNAs with shorter 3′ UTRs (Flavell et al, 2008; Sandberg et al, 2008; Ji et al, 2009; Mayr & Bartel, 2009; Shepard et al, 2011; Elkon et al, 2012). In contrast, some differentiated or quiescent cells tend to use downstream or distal PAS to produce mRNAs with longer 3′ UTRs (Flavell et al, 2008; Sandberg et al, 2008; Ji et al, 2009; Mayr & Bartel, 2009; Shepard et al, 2011; Elkon et al, 2012). These correlations implicate APA regulation in cellular fate specification. However, it remains unclear whether global APA changes are mere consequences of upstream gene regulation events or APA regulation plays a direct role in modulating differentiation and development. It should be pointed out that the relationship between the global APA profile and the cellular proliferation/differentiation status may be more complicated and subject to tissue-specific regulation (Lianoglou et al, 2013).
Despite widespread APA regulation in many important physiological processes, it remains poorly understood how APA is regulated. Mammalian mRNA 3′ processing typically involves an endonucleolytic cleavage and polyadenylation and requires the poly(A) polymerase (PAP) and four protein complexes, including the cleavage and polyadenylation specificity factor (CPSF), the cleavage stimulation factor (CstF), cleavage factor I (CF Im) and II (CF IIm) (Zhao et al, 1999; Chan et al, 2011). CPSF, CstF, and CF Im are involved in PAS recognition by binding to the AAUAAA hexamer, the downstream U/GU-rich element, and UGUA-containing auxiliary elements, respectively (Zhao et al, 1999; Chan et al, 2011). Several mRNA processing factors have been shown to regulate APA (Jenal et al, 2012; Martin et al, 2012; Yao et al, 2012). However, it remains unclear how these general 3′ processing factors modulate the APA of a specific set of transcripts and why they have distinct effect on the APA patterns in different mRNAs. Therefore, systematic analyses are needed to understand how the core mRNA 3′ processing factors regulate APA individually and how their actions are coordinated.
Embryonic stem cells (ESCs) are not only an important model system for understanding the fundamental mechanisms of development, but also hold great promise for regenerative medicine, disease modeling, and drug discovery (Murry & Keller, 2008). ESCs have two defining properties: the ability to become any other cell type of the three germ layers, known as pluripotency, and the ability to proliferate indefinitely while maintaining the pluripotent state, known as self-renewal (Smith, 2001). Currently, our understanding of ESC self-renewal has been largely restricted to transcriptional regulation and chromatin dynamics (Ng & Surani, 2011; Young, 2011). What has not been addressed is whether post-transcriptional mechanisms, especially APA, provide another layer of control. The observations of widespread APA changes and 3′ UTR lengthening during stem cell differentiation and early embryonic development (Ji et al, 2009; Shepard et al, 2011; Boutet et al, 2012), together with 3′ UTR shortening during somatic reprogramming to iPSCs (Ji & Tian, 2009), suggest that APA is tightly regulated during cell fate transitions and may play critical roles in stem cell biology. Thus, it is of great interest to identify key APA regulators in stem cells and characterize their functional impact.
In this study, we identify Fip1, a component of the CPSF complex, as a critical APA regulator in ESCs. We show that Fip1 promotes ESC self-renewal and somatic cell reprogramming, in part, by activating ESC-specific APA patterns and in turn the expression of critical target genes. Furthermore, we have characterized the mechanism for Fip1-mediated APA regulation and its target specificity. Together, our study demonstrates an important role for APA regulation in stem cell functions and cell fate determination.
Results
The mRNA 3′ processing factor Fip1 is required for ESC self-renewal and pluripotency
Fip1, encoded by the Fip1 l1 gene, is an essential mRNA 3′ processing factor that is conserved from yeast to human (Kaufmann et al, 2004). Mammalian Fip1 is a subunit of CPSF and is involved in PAS recognition and in recruiting PAP to the 3′ processing complex (Kaufmann et al, 2004). Interestingly, Fip1 has recently been identified in genome-wide RNAi screens as a potential self-renewal factor in mouse ESCs (Ding et al, 2009; Hu et al, 2009). To examine its role in self-renewal, we depleted Fip1 in mouse ESCs using two independent siRNAs. Knock-down (KD) resulted in greater than 70% reduction in Fip1 at both the mRNA and protein levels, without significantly affecting the protein levels of other CPSF subunits (Fig 1A and Supplementary Fig S1). With an ESC reporter line (Oct4GiP), in which the expression of the enhanced green fluorescent protein (EGFP) is driven by the ESC-specific Oct4 promoter (Ying et al, 2002; Zheng & Hu, 2012), we found that Fip1 KD led to impaired ESC maintenance and identity, as determined by the loss of EGFP expression in a significant portion of cells (Fig 1B and Supplementary Fig S2). These results demonstrated an essential role for Fip1 in ESC self-renewal. Further supporting this conclusion, Fip1 KD in both Oct4GiP and J1 ESCs resulted in the loss of the stereotypical ESC colony morphology and a decrease in alkaline phosphatase (AP) staining (an ESC marker) (Fig 1C and Supplementary Fig S3A and B), signifying the loss of ESC identity and exit from self-renewal. Colony formation assays showed that Fip1 KD led to a significant increase in the percentage of partially differentiated and differentiated cells (Supplementary Fig S3C, P = 0, Pearson's chi-square test) and significantly lower number of undifferentiated ESC colonies (Supplementary Fig S3D, P < 0.01). At the molecular level, reverse transcription and quantitative polymerase chain reaction (RT–qPCR) analyses detected gene expression changes for important marker genes after Fip1 KD, including decreases in ESC markers such as Oct4, Nanog, Sox2, Esrrb, Rex1, Lefty1, and Lefty2, as well as increases in differentiation markers including Eomes, Sox17, Foxa1, Gata3, Kdr, Col5a2, and Gcm1 (Fig 1D). Finally, like other self-renewal factors (Wang et al, 2012), we found that Fip1 KD led to aberrant expression of lineage markers during ESC differentiation by embryoid body (EB) formation, including enhanced up-regulation of Sox1, Sox17, Gata6, Kdr and reduced up-regulation of Fgf5 early on (day 2) and reduced up-regulation of all the markers tested at later time points (days 4 and 6) (Supplementary Fig S4). These results indicate that Fip1 also plays an important role in governing the developmental potential of ESCs. Therefore, we conclude that Fip1 is required for ESC self-renewal and pluripotency. Consistently, we found that the protein levels of Fip1 and several other CPSF subunits decrease during ESC differentiation triggered by LIF (leukemia-inducing factor) withdrawal (Fig 1E and Supplementary Fig S5). Thus, Fip1 expression is developmentally regulated.
Figure 1. Fip1 is required for ESC self-renewal.

A Fip1 knock-down in ESCs. Western blots of Fip1 and other CPSF subunits using cell lysates prepared from ESCs 4 days after transfection with lipids only (Mock), control siRNA (siControl), or Fip1 siRNAs (siFip1-2 and x-3).
B–D Impact of Fip1 KD on ESC self-renewal. OctGiP ESCs were transfected with the indicated siRNAs, and cells were analyzed 4 days post-transfection by FACS (the percentages of GFP-negative cells are plotted as mean ± s.e.m. [n = 3; **P < 0.01)] (B), or by AP staining (C), or by RT–qPCR to determine lineage marker expression [expression values are normalized to mock and plotted as mean ± s.e.m. (n = 3)] (D). Marker genes that showed a significant different expression between siControl and siFip1-2/3 are marked by * (P < 0.05).
E ESCs were cultured without LIF for 0, 2, 3, and 4 days and harvested for Western analyses of specified factors.
Fip1 regulates mRNA alternative polyadenylation in ESCs
We next characterized the mechanisms by which Fip1 promotes ESC self-renewal. Given the known function of Fip1 in mRNA 3′ processing and the distinct APA signatures for ESCs and differentiated cells (Ji & Tian, 2009; Shepard et al, 2011), we hypothesized that Fip1 promotes ESC self-renewal, at least in part, by regulating APA. To test this hypothesis, we determined the impact of Fip1 on the global APA profile in ESC by direct RNA sequencing (DRS) analysis of ESCs transfected with control or Fip1-specific siRNAs. DRS using the Helicos platform maps RNA polyadenylation sites globally with nucleotide resolution and is highly quantitative as no library preparation is required (Ozsolak et al, 2010). Indeed, when the gene expression profiling results of control ESC and Fip1 KD samples based on DRS and microarray were compared, they showed excellent agreement (r2 =0.94) (Supplementary Fig S6). Based on the DRS data, we have identified 374 genes that showed significantly different APA profiles upon Fip1 depletion in ESCs (Fisher's exact test, FDR < 10−4) (Fig 2A). In 311 or 83% of these genes, there was a relative increase in the mRNAs polyadenylated at the distal PASs in Fip1 KD cells (P < 10−40, binomial test) (Fig 2A). This type of APA change, referred to as proximal-to-distal (PtoD) shift, leads to 3′ UTR lengthening when both PASs are in the same exon. Examples of the APA changes induced by Fip1 depletion are shown in Fig 2B and Supplementary Fig S7. The fold change of these and other APA events identified by DRS analysis was validated by RT–qPCR assays (Supplementary Fig S8). These results suggest that Fip1 regulates the APA of a specific set of genes in ESCs and, in most cases, promotes the usage of proximal PASs and production of mRNAs with shorter 3′ UTRs. Interestingly, our gene expression analyses revealed that only seven of the 311 Fip1 PtoD APA target genes showed significant changes in total mRNA levels after Fip1 KD (> twofold, FDR < 0.01, Supplementary Table S1), suggesting that Fip1 regulates the APA profiles of its target genes without significantly affecting their transcript levels.
Figure 2. Fip1 regulates the alternative polyadenylation (APA) of potential self-renewal factors in ESCs.

- Direct RNA sequencing analysis of APA in ESCs 4 days after they were transfected with siControl or siFip1-2. Log2 (proximal/distal ratio) are plotted for ESCs transfected with control siRNA (y-axis) and siFip1-2 (x-axis). Statistically significant (P < 10−4, Fisher's exact test) changes are colored in blue (PtoD: proximal-to-distal shift) or red (DtoP: distal-to-proximal shift). The numbers of PtoD and DtoP APA changes are shown in a column graph (inset).
- UCSC genome browser tracks showing the Direct RNA sequencing results for the Ahctf1 and Ncaph2 genes in ESC with control (top track) or Fip1 KD (bottom track).
- The expression levels of all genes (left panel) or Fip1 PtoD APA genes (right panel) during ESC embryoid body (EB) differentiation time course (0–96 h, x-axis). Genes are ordered based on their expression levels in ESCs, and the standardized expression values are plotted as a heat map.
- Gene ontology analysis of 311 PtoD Fip1 APA targets using Ingenuity © analysis. Functional categories (y-axis) and the corresponding P-values (x-axis) are shown.
- A Venn diagram showing the overlap between APA changes induced by Fip1 KD and those observed during ESC differentiation into neurons. The blue circles are PtoD genes and the red DtoP. The numbers of genes in each area are marked.
To assess whether the impaired ESC maintenance observed in Fip1 KD cells (Fig 1) is the cause or consequence of APA modulation, we analyzed APA and lineage markers at earlier time points, including 24 and 48 h after Fip1 siRNA transfection. Depletion of Fip1 protein and APA changes in Fip1 targets were detected as early as 24 h after siRNA transfection (Supplementary Figs S9 and S10A). In contrast, no obvious differentiation has occurred at this point based on cell morphology (Supplementary Fig S9) and ESC and differentiation marker expression (Supplementary Fig S10B). Together, these data suggest that Fip1 regulates the APA of a specific set of transcripts in ESCs and that APA changes take place prior to the loss of ESC identity following Fip1 depletion.
Fip1-mediated APA regulation modulates the expression of self-renewal factors
To determine whether and how Fip1-mediated APA regulation contributes to ESC self-renewal, we next examined the functions of Fip1 APA target genes. Multiple lines of evidence suggest that Fip1 APA target genes have important roles in ESCs. First, Fip1 APA genes are highly expressed in ESCs but are down-regulated during differentiation by EB formation (P < 2.2 × 10−16 at both 48 and 96 h, Fig 2C and Supplementary Fig S11), indicating that these genes have ESC-specific functions. In addition, these results also suggest that Fip1 APA genes are regulated at both transcriptional and post-transcriptional levels (such as APA) during differentiation. Second, gene ontology analysis of the Fip1 PtoD APA targets revealed a significant enrichment of genes that function in cell cycle, proliferation, and development (Fig 2D). Third, ˜25% of the PtoD APA changes induced by Fip1 depletion in ESCs were observed during ESC differentiation into neurons (P < 10−40) (Fig 2E) (Shepard et al, 2011). In 86% of these genes, polyadenylation shifted to the same distal PASs during neuronal differentiation and in Fip1-depleted ESCs. In the majority of the remaining 14% of genes, there are three or more PASs and polyadenylation shifted to multiple distal PASs. We have also examined the APA profiles of eight Fip1 targets during retinoic acid-induced neuronal differentiation by RT–qPCRs and found that four targets (50%) showed PtoD APA shifts during this process, similar to what was observed in Fip1 KD cells (Supplementary Fig S12). Finally, 16 of the Fip1 APA targets were identified in previous genome-wide RNAi screens as potential ESC self-renewal factors (Ding et al, 2009; Hu et al, 2009) (Supplementary Table S2). These observations suggest that many Fip1 APA targets may function in ESC self-renewal and their APA patterns are developmentally regulated.
As Fip1 depletion in ESCs leads to 3′ UTR lengthening in the majority of its target genes, we next examined the impact of 3′ UTR extension on the expression of these genes. To this end, we selected a panel of Fip1 APA target genes, including Ahctf1, Ino80e, Ncl, Nfyb, Rbx1, Sbno1, Wdr18, Etf1, Wwp2, and Ncaph2, all of which displayed 3′ UTR lengthening in Fip1 KD cells (Fig 2B and Supplementary Fig S7). We next cloned their constitutive 3′ UTRs (cUTRs or the 3′ UTRs of mRNAs polyadenylated at the proximal PAS) or the 3′ UTRs of the longer APA isoform that contain both cUTR and alternative 3′ UTRs (aUTRs) downstream of a firefly luciferase reporter gene (Fig 3A, left panel). A heterologous SV40 PAS was used to terminate all transcripts to ensure equal 3′ processing efficiency. When equal molar amounts of the reporter constructs were transfected into ESCs, the cUTR plus aUTRs (c+aUTRs) resulted in lower luciferase expression for nine out of the eleven Fip1 target genes tested compared to the cUTRs (Fig 3A, right panel), suggesting that Fip1 KD-induced 3′ UTR extension suppresses protein expression. As a control, we also tested the anti-sense sequences of the c+aUTRs from four of the Fip1 targets in the same assay and found that at least two of them had significantly different effects on reporter gene expression compared to their corresponding c+aUTRs (Supplementary Fig S13), suggesting that the inhibitory effect of aUTRs may be due to their lengths and/or sequences. In keeping with the reporter assay results, we detected decreases in the endogenous protein levels of several Fip1 APA target genes in Fip1 KD cells, including Ncaph2, Wwp2, and Etf1 (Fig 3B, quantification in Supplementary Fig S14). These data strongly suggest that the Fip1 depletion-induced 3′ UTR lengthening leads to gene silencing in ESCs.
Figure 3. Fip1-mediated alternative polyadenylation (APA) regulation modulates the expression of self-renewal factors in ESCs.

- Luciferase reporter assays to determine the impact of the cUTRs (in blue) or c+aUTR (in green) on gene expression. ESCs were transfected with a reporter construct containing the firefly luciferase (Fluc) coding sequence linked to the specified 3′ UTRs and a control Renilla luciferase (Rluc) construct, and the Fluc/Rluc ratio was determined 2 days post-transfection. The Fluc/Rluc ratio (relative expression, y-axis) is normalized to cUTR-containing construct and plotted as mean ± s.e.m. (n = 3). n.s.: not significant; *P < 0.05.
- Western blot analyses of the Fip1 APA targets using lysates prepared from ESCs 4 days after they were transfected with the specified siRNAs.
- Fip1 APA target genes are required for ESC self-renewal. ESCs were transfected with specified siRNAs and cellular morphology was imaged 4 days post-transfection. A second siRNA was used to deplete each gene in ESC and the cell images are shown in Supplementary Fig 15B.
To directly test the functions of Fip1 APA target genes in ESC self-renewal, we silenced a number of Fip1 APA target genes in ESCs by RNAi using two distinct siRNAs for each target gene (Fig 3C and Supplementary Fig S15). Strikingly, similar to Fip1 KD, depletion of these factors, including Ahctf1, Ncl, Rcc2, Rbx1, Wdr18, Ncaph2, Wwp2, and Etf1, resulted in impaired ESC maintenance as characterized by changes in cell morphology and the aberrant expression of ESC and differentiation markers (Fig 3C and Supplementary Figs S15B and S16). The depletion of these factors led to changes in different marker genes (Supplementary Fig S15), indicating that KD of these factors induces ESC differentiation into different lineages. As controls, we have also knocked down Apex1 and Nhp2, two highly expressed genes in ESC that did not show significant APA changes upon Fip1 KD. Depletion of these two factors did not result in the ESC differentiation and loss of self-renewal (Supplementary Fig S15), demonstrating the specificity of our assays. Taken together, these data suggest that Fip1 promotes ESC self-renewal by maintaining short 3′ UTRs and thereby the optimal expression of critical self-renewal factors.
Fip1 is required for somatic cell reprogramming
Many known ESC self-renewal regulators also have important roles in the establishment of the pluripotent state (Orkin & Hochedlinger, 2011). Therefore, we asked whether Fip1 is required for the generation of induced pluripotent stem cells (iPSCs) from mouse embryonic fibroblasts (MEFs). During the course of reprogramming, we observed an increase in Fip1 mRNA levels (Fig 4A). Importantly, we also detected concomitant 3′ UTR shortening for several Fip1 APA target genes (Fig 4B), suggesting that the ESC-specific APA profile is restored during reprogramming. Consistent with this observation, 40 Fip1 APA target genes were previously reported to show 3′ UTR shortening during reprogramming (Ji & Tian, 2009) (P < 10−15, Supplementary Table S3). These observations suggest that the Fip1 APA program is developmentally regulated and that Fip1 may participate in somatic reprogramming. To test this possibility, we silenced Fip1 in MEFs using a lentiviral-based shRNA (Supplementary Fig S17A). In contrast to its inhibitory effect on the self-renewal and proliferation of ESCs (Fig 4C, left panel and Fig 1), Fip1 depletion had little, if any, effect on cell growth or viability in MEFs (Fig 4C, right panel). Remarkably, however, Fip1 KD in MEFs caused a dramatic reduction in their reprogramming efficiency as determined by AP staining (Fig 4D). Fip1 KD also led to an increase in aUTR usage during reprogramming (Supplementary Fig S17B), but its effect was more modest compared to that in ESCs. To more accurately quantify the impact of Fip1 on reprogramming efficiency, we used MEFs that harbor a reporter EGFP gene under the control of the Oct4 promoter for reprogramming (Brambrink et al, 2008), and found that Fip1 silencing caused an 88-fold reduction in the number of EGFP-positive iPSCs (Fig 4E). To understand what stage of reprogramming is affected by Fip1 KD, we determined the expression of early iPSC markers at day 3 and day 6. We detected lower induction of iPSC markers as early as day 3, while expression of the cell cycle marker Cdkn2b was not affected (Supplementary Fig S18). Thus, Fip1 is required for successful reprogramming of somatic cells into iPSCs from an early stage and may also contribute to iPSC maintenance.
Figure 4. Fip1 is required for somatic reprogramming.

- RT–qPCR analyses of Fip1 levels (x-axis) at the specified time during reprogramming (y-axis). Fip1 levels are normalized to day 0 level and plotted as mean ± s.e.m. (n = 3).
- Alternative polyadenylation (APA) changes of Fip1 targets during reprogramming. RT–qPCR analyses of aUTR usage in Fip1 APA genes at the specified time during reprogramming. aUTR usage values are normalized to the day 0 value and plotted as mean ± s.e.m. (n = 3).
- The growth curves of ESCs (left) or MEFs transfected with control (blue line) or Fip1 si/shRNA (red line).
- Impact of Fip1 silencing on somatic cell reprogramming. The number of AP+ colonies was counted 12 days after the introduction of Oct4, Sox2, Klf4, and Myc by lentivirus transduction of MEF. Top panel: the AP staining image of MEFs induced to reprogram after transfected with control or Fip1-shRNA. Bottom panel: quantification results of APA-positive colonies are plotted as mean ± s.e.m. (n = 3).
- Reprogramming efficiency was determined by the percentage of Oct4GFP-positive cells on day 12 by FACS analysis. GFP-positive cells are boxed (Top panel). Bottom panel: Quantification results of GFP-positive iPSCs based on the FACS data are plotted as mean ± s.e.m. (n = 3). **P < 0.01.
Fip1 controls the activity of the mRNA 3′ processing machinery in ESCs
Fip1 KD leads to a shift to distal PASs and 3′ UTR lengthening in most of its target mRNAs (Fig 2A). To define the underlying mechanism, we first examined the effect of Fip1 depletion on the overall mRNA 3′ processing activity. Rpl26 encodes a ribosomal protein that is highly expressed in mouse ESCs, and its mRNAs are polyadenylated at a dominant PAS in ESCs (Fig 5A). We estimated the cleavage/polyadenylation efficiency at Rpl26 PAS by measuring the ratio between the uncleaved and total transcripts via RT–qPCR (Fig 5A). Our analysis detected a significant increase in the percentage of uncleaved Rpl26 transcripts upon Fip1 depletion (Fig 5A), suggesting lower cleavage/polyadenylation efficiency at this PAS in Fip1 KD cells. To determine whether Fip1 impacts additional PASs in a similar manner, we measured the cleavage/polyadenylation efficiencies for a number of Fip1-regulated PASs in ESCs using a bicistronic luciferase reporter assay (Yao et al, 2012) (Supplementary Fig S19A). In this reporter system, efficient 3′ processing at the tested PAS only allows the expression of the Renilla luciferase (Rluc) gene, while poor 3′ processing efficiency would lead to transcription read-through and the expression of both Rluc and the downstream firefly luciferase (Fluc) gene. Thus, the Rluc/Fluc ratio reflects the cleavage/polyadenylation efficiency at the tested PASs. Using this assay, we found that Fip1 depletion in ESCs resulted in lower 3′ processing activities at all 12 PASs tested (Supplementary Fig S19B). To more directly determine the effect of Fip1 depletion on mRNA 3′ processing, we carried out in vitro coupled cleavage/polyadenylation assays or cleavage assays using the nuclear extract from control or Fip1 KD cells and RNA substrates containing the L3 PAS, a well-characterized viral PAS (Shi et al, 2009). A significant decrease in the mRNA 3′ processing efficiency was observed for nuclear extract prepared from Fip1 KD cells (Fig 5B). Therefore, both our in vivo and in vitro data strongly suggest that Fip1 KD leads to lower mRNA 3′ processing activities and more transcription read-through. On the other hand, our microarray analyses demonstrated that the mRNA levels of the vast majority of the genes (˜98%) were unaltered in Fip1 KD cells, suggesting that the decrease in 3′ processing activity was not sufficient to cause a general defect in gene expression (further discussed below).
Figure 5. Mechanisms for Fip1-mediated alternative polyadenylation (APA) regulation.

- Fip1 controls mRNA 3′ processing activity in vivo. Top panel: UCSC Genome Browser track showing the 3′ end of Rpl26 mRNA and the positions of the RT–qPCR primers. Bottom panel: Relative transcription read-through is measured by the ratio between uncleaved (amplified by forward and reverse 2 primers) and the total transcripts (forward and reverse 1 primers) based on RT–qPCR results, and normalized to the value in siControl and plotted as mean ± s.e.m. (n = 3).
- In vitro cleavage/polyadenylation (top) and cleavage (bottom) assays using L3 RNA substrate and nuclear extract from mock or Fip1 KD cells. Pre-mRNAs, poly(A)+, and 5′ cleavage products are marked.
- PAS activities from the proximal (blue) or distal (green) PAS of the specified genes (x-axis) were determined by transfecting pPASPORT constructs into ESCs and measuring the Rluc/Fluc ratio 1 day after transfection, normalized by the value of the distal PASs, and plotted as mean ± s.e.m.
- Distribution of Fip1 binding sites (blue line, based on iCLIP-seq data) and cleavage sites (green line, based on DRS data) relative to the closest upstream A(A/U)UAAA. Position 0 represents the 5′ end of AAUAAA.
- Fip1 iCLIP maps for the cUTR and aUTRs of PtoD (blue), DtoP (red), and non-target genes (gray). Fip1 iCLIP signals are first normalized by transcript numbers (DRS read counts). cUTRs and aUTRs are divided into 100 bins each, and the summation of iCLIP signals in each bin for all genes in the group is divided by the number of genes. The normalized iCLIP signals (y-axis) are plotted for cUTRs and aUTR regions (x-axis). The difference in the density of the iCLIP peaks between PtoD and DtoP genes is likely due to the different numbers of genes in these groups.
- A box plot showing the distribution of the distances between the proximal and distal PASs (within the same exons) for PtoD, DtoP, and non-target genes. The red lines mark the median values: 987 nt for PtoD genes, 205 nt for DtoP genes, and 759 nt for non-targets.
To understand how a decrease in mRNA 3′ processing activity caused by Fip1 KD can lead to PtoD APA changes, we analyzed the sequence (−100 nt to +100 nt relative to the cleavage site) of the PAS pairs that showed PtoD APA changes in Fip1 KD cells. We focused on this region because it contains all the known key cis-elements for mRNA 3′ processing and is the most highly conserved region in both the proximal and distal PASs in Fip1 APA targets (Supplementary Fig S20). We found that AAUAAA and UG-rich motifs, two key cis-elements that define the canonical mammalian PASs (Chan et al, 2011), are more frequently found at the distal PASs than at the proximal sites (Supplementary Fig S21A). To determine whether these sequence features make the distal sites stronger than the proximal ones, we used two different assays to compare the proximal and distal PAS pairs from Fip1 target genes. First, using the bicistronic luciferase reporter construct described earlier (Supplementary Fig S19A), we found that the distal PASs are significantly more active than the corresponding proximal sites for all Fip1-regulated PAS pairs tested (sequences from −100 nt to +100 nt were used, Fig 5C). This result was further confirmed by in vitro cleavage/polyadenylation assays with proximal and distal PASs from Fip1 target genes (Supplementary Fig S21B). These data strongly suggest that the distal PASs for Fip1 target genes are intrinsically stronger than the proximal sites as they contain more canonical sequence features in their core region and thus have higher affinity for the 3′ processing machinery. As Fip1 is expressed at higher levels in ESCs (Fig 1E), our results support a model in which high levels of Fip1 promote the efficient recognition and processing at the suboptimal proximal PASs in ESCs. Fip1 depletion impairs proximal PAS usage and leads to higher usage of the stronger distal PASs by the 3′ processing machinery. This model extends a previously proposed model for CstF64-mediated APA regulation in B cells, referred to as the “survival of the fittest” model (Takagaki et al, 1996; Shi, 2012), and explains the directionality of the majority of APA changes induced by Fip1 depletion.
Fip1 directs the specificity of APA site usage in ESCs
To understand how Fip1 regulates the APA of a specific set of self-renewal genes, we mapped Fip1-RNA interactions on the transcriptome levels in ESC by individual nucleotide resolution CLIP and high-throughput sequencing (iCLIP-seq) analyses (Konig et al, 2010). Our iCLIP-seq analysis showed that Fip1 preferentially binds to U-rich sequences upstream of the AAUAAA hexamer and the cleavage/polyadenylation sites in vivo (Fig 5D). Based on the transcriptome-wide iCLIP data, we have generated a Fip1-RNA interaction map for Fip1-regulated APA events by plotting the normalized iCLIP signals in the cUTRs or the aUTRs (Fig 5E) (see Materials and Methods for details). Significantly higher Fip1 iCLIP signals were detected in the 3′ UTRs of Fip1 APA target mRNAs than non-target mRNAs (P = 1.1 × 10−18, Fig 5E). Therefore, Fip1 APA target and non-target genes appear to differ in Fip1-RNA interaction strengths and/or frequencies. Additionally, a comparison between the proximal PASs of Fip1 APA targets and non-targets detected enrichment of canonical PAS features including AAUAAA and UG-rich downstream elements for Fip1 APA targets (Supplementary Fig S22). These results implicate Fip1-RNA interactions and PAS sequences in determining the specificity of Fip1-mediated APA regulation.
Finally, we wanted to understand how Fip1 depletion induces PtoD APA changes in some targets but the opposite shifts in others (Fig 2A). In searching for sequences or molecular features that may explain the different modes of Fip1-mediated APA regulation, we found a significant difference in the distance between the proximal and distal PASs in these two groups of genes. For example, Dicer1 and Hspa4 are two representative PtoD genes and the distance between their proximal and distal PASs is 3,697 nt and 1,756 nt, respectively (Supplementary Fig S23A). In contrast, for DtoP genes Arl6ip1 and Ctps, the distance between the proximal and distal PASs is only 74 nt and 285 nt, respectively (Supplementary Fig S23B). Indeed, when we compared all PtoD and DtoP genes whose proximal and distal PASs are in the same terminal exons, the median distance between their alternative PASs is 987 nt for PtoD genes, but only 205 nt for DtoP genes (P < 10−5, K-S test) (Fig 5F). Such a significant difference may affect how Fip1 impacts the choice between alternative PASs. Together, these results suggest that the specificity and the mode of Fip1-mediated APA regulation are determined, at least in part, by Fip1-RNA interactions and the context of the alternative PASs. These data have been incorporated in a mechanistic model for Fip1-mediated APA regulation as described below (Fig 6).
Figure 6. A model for Fip1-mediated alternative polyadenylation (APA) regulation.
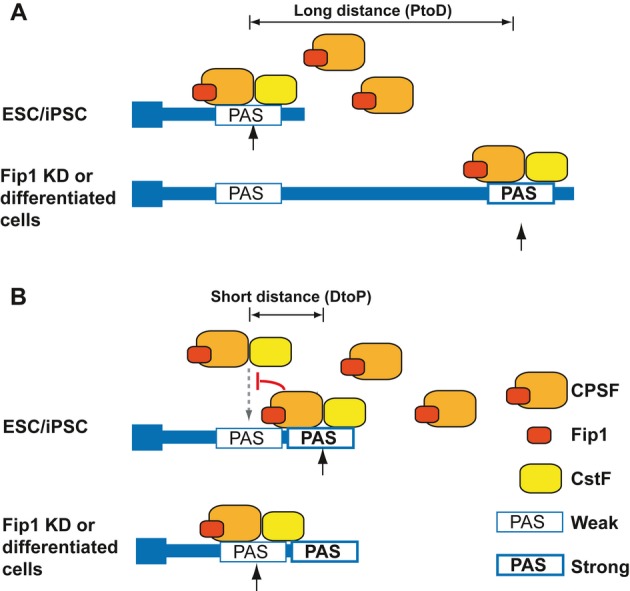
- For APA genes whose proximal and distal PASs are far from each other, there is a significant lag between the times when the alternative PASs are transcribed. Higher levels of Fip1/CPSF (such as in ESCs and iPSCs) promote the recognition of weaker proximal PASs. When Fip1/CPSF levels are low, transcription read-through the usage of stronger distal PASs increases.
- For APA genes whose proximal and distal PASs are close to each other, these alternative PASs directly compete for binding to mRNA 3′ processing factors. At high levels, Fip1/CPSF binding to the region between the two PASs may block CstF binding in the same region and the recognition of the proximal PAS. At lower levels, Fip1/CPSF binding decreases, which allow CstF binding to this region, leading to better recognition of the proximal PAS. See Discussion for more details.
Discussion
Although widespread APA changes have been detected in development (Di Giammartino et al, 2011; Shi, 2012; Elkon et al, 2013; Mueller et al, 2013; Tian & Manley, 2013), the functional significance of APA in regulating cell growth/differentiation is not well understood. In this study, we demonstrated that Fip1 is essential for ESC self-renewal and somatic cell reprogramming (Figs1 and 4). Fip1 maintains the ESC-specific APA profiles and promotes the production of mRNAs with shorter 3′ UTRs for a subset of genes (Fig 2). Such APA profiles are important for the optimal expression of Fip1 target genes, many of which encode critical ESC self-renewal factors (Fig 3). These results suggest that Fip1 promotes ESC self-renewal, at least in part, by maintaining the APA profile and in turn the optimal expression of many self-renewal factors. Thus, our study provided, to our knowledge, the first experimental evidence that APA regulation directly contributes to cell fate determination. Additionally, we showed that the specificity and the mode of Fip1-mediated APA regulation are dependent on Fip1-RNA interactions and the context of alternative PASs. These results may have broad implications not only for our understanding of the mechanisms and functional significance of APA regulation, but also for the role of post-transcriptional gene regulation in stem cell biology and cell fate specification.
Based on our data, we propose the following model for Fip1-mediated APA regulation (Fig 6). For genes whose alternative PASs are distant from one another (Fig 6A), there is a significant lag between the times when the proximal and the distal PASs are transcribed. When Fip1 and the mRNA 3′ processing activities are high such as in ESCs, cleavage/polyadenylation occurs at the weaker proximal PASs before the distal PAS is transcribed or recognized. However, when mRNA 3′ processing activity is low, the efficiency of cleavage/polyadenylation at the proximal PASs decreases and transcription read-through increases, which allows the transcription of the stronger distal PASs that are preferentially recognized by the mRNA 3′ processing machinery due to their higher intrinsic strengths. For genes whose alternative PASs are close to one another (Fig 6B), the distal sites are transcribed immediately after the proximal sites and these sites compete for binding to the mRNA 3′ processing factors. When Fip1 levels are high, the distal PASs are favored due to their higher intrinsic strengths. Given the short distance between the PASs, the region between the two alternative PAS may serve both as DSEs (thus CstF-binding site) for the proximal PAS and as Fip1/CPSF-binding site for the distal PAS. Thus, Fip1/CPSF binding in this region may directly interfere with CstF binding and the recognition of the proximal PASs, leading to their repression. At lower Fip1 levels, a decrease in Fip1/CPSF binding in this region allows increased CstF recruitment and consequently better recognition of the proximal PASs. This model extends the classical “survival of the fittest” model by incorporating the distance between alternative PASs as a new parameter, and explains how Fip1, and possibly other mRNA 3′ processing factors, can differentially regulate APA on different transcripts. Additionally, since the 3′ UTR differences are significantly greater for the PtoD genes, Fip1-mediated APA regulation is likely to have a greater impact on the expression of the PtoD genes than that of the DtoP genes.
Although our study has focused on the effect of Fip1-mediated APA regulation on ESC self-renewal, we have also evaluated the impact of Fip1 on single-PAS genes. Of 5,947 single-PAS genes (based on high-confidence DRS peaks with 10 reads or more), only 77 genes or 1.3% showed a significant decrease in mRNA levels in Fip1 KD cells based on microarray data (by 50% or more, FDR < 0.05). Gene ontology analyses failed to detect any enrichment of functional groups in these genes (Supplementary Fig S24A). The PASs of these 77 genes in general contain less canonical polyadenylation features, including AAUAAA and the UG-rich downstream element (Supplementary Fig S24B), and have significantly less Fip1 iCLIP signals (Supplementary Fig S25A). No significant difference in the expression levels was detected between these 77 genes and others (Supplementary Fig S25B). Together, these results suggest that Fip1 KD did not cause a general mRNA 3′ processing defect and Fip1 seems to play a very limited role in regulating single-PAS genes under the conditions used in this study. To understand why the polyadenylation pattern of single-PAS genes did not change when Fip1 KD led to decreased PAS recognition (Fig 5A, B), we searched for potential PAS in the 5-kilobase region downstream of the mapped PAS using a previously published support vector machine (SVM)-based method (Cheng et al, 2006). Our results suggest that the potential downstream PASs are significantly weaker than the mapped PASs of both single-PAS and APA genes based on the SVM scores (Supplementary Fig S26). These results suggest that, in contrast to APA genes, polyadenylation did not change for single-PAS genes following Fip1 KD because there is no stronger PAS downstream.
In addition to Fip1, it is likely that other mRNA 3′ processing factors may play an important role in ESCs as well. In fact, CPSF73 was also identified as a potential self-renewal factor in previous genome-wide RNAi screens (Ding et al, 2009; Hu et al, 2009). Consistently, we showed that the protein levels of CPSF73 and other CPSF subunits are regulated in a similar manner as Fip1 during differentiation (Fig 1E). However, not all mRNA 3′ processing factors have equally important functions in self-renewal. For example, when we depleted CPSF30 or CF Im25 in ESCs by RNAi (Supplementary Figs S27–28), we did not detect the same APA changes as those induced by Fip1 KD (Supplementary Fig S28A). More importantly, no obvious loss of self-renewal was observed based on cell morphology (Supplementary Fig S27B) or marker gene expression (Supplementary Fig S28B). It will be of interest to systematically characterize the functions of other mRNA 3′ processing factors and APA regulators in ESCs and development. Interestingly, the mRNA 3′ processing factors and APA regulators seem to have cell type-specific effect on cell proliferation. For example, Fip1 KD led to an impaired proliferation of ESCs, but had no effect on the growth of MEF or HeLa cells (Fig 5B, Supplementary Fig S29B) even though Fip1 KD did cause predominantly PtoD APA changes in HeLa cells (Supplementary Fig S29A and C).
Several core mRNA 3′ processing factors, including PABPN1, CF Im68, CstF64, and CstFτ, have been shown to regulate APA globally (Jenal et al, 2012; Martin et al, 2012; Yao et al, 2012). To begin to understand APA regulation mediated by the core 3′ processing factors at the systems level, we have compared the target specificity and the directionality of APA regulation mediated by Fip1 and other aforementioned 3′ processing factors. When we directly compared the genes whose APA is regulated by Fip1 (this study) and those by PABPN1 (Jenal et al, 2012) or CstF64&τ-RNAi (Yao et al, 2012) or CF Im68 (Martin et al, 2012), relatively little overlap was found (Supplementary Fig S30A–C). Therefore, although these factors are believed to play a general role in mRNA 3′ processing, each seems to regulate the APA of a specific set of transcripts. It should be pointed out that such differences could also be due, at least in part, to the fact that these studies were carried out in different cell types or species. In addition to target specificity, the directionality of APA regulation by these factors also differs significantly. For examples, depletion of Fip1 or CstF64/τ leads to mostly PtoD APA shifts while knocking down CF Im or PABPN1 shifts the APA profiles in the opposite direction (Supplementary Fig S30D). Therefore, these core 3′ processing factors appear to regulate APA through very different mechanisms and it requires more systematic studies to understand both their target specificity and directionality.
Finally, our results identified APA as a post-transcriptional mechanism that plays a critical role in ESCs, providing a new layer of control on ESC self-renewal and pluripotency. While transcription may function as on-off switches in gene regulation in ESCs (Ng & Surani, 2011; Young, 2011), Fip1-mediated APA regulation modulates gene expression without significantly impacting the mRNA levels (Supplementary Fig S14A and Table S1). Here, we show that 3′ UTR extension suppresses protein expression for Fip1 target mRNAs, possibly due to the presence of binding sites for miRNAs and/or RNA-binding proteins in the extended UTRs (Sandberg et al, 2008; Mayr & Bartel, 2009), which in turn may affect mRNA stability and/or translation. Indeed, the number of predicted microRNA target sites in the a+cUTR regions is 2.5 times of that in the cUTRs for Fip1 APA target genes although the density of miRNA target sites seems to be lower in the aUTRs (Supplementary Fig S31). We propose that, similar to microRNA-mediated gene silencing (Bartel, 2009), APA regulation serves as a fine-tuning mechanism for gene regulation in ESCs. Although the effect of APA changes on individual genes may be modest, its cumulative effect on a group of functionally related genes can have significant consequences on cellular state. Our study provides one example of how APA regulation can influence a physiological process. Given the widespread APA changes across the immune and neural systems, embryonic development, and oncogenesis (Di Giammartino et al, 2011; Shi, 2012), APA regulation may also contribute to cellular transitions in other systems.
Materials and Methods
ESC culture and transfections
E14Tg2a (E14) and J1 cells were obtained from Mutant Mouse Research Resource Centers and American Type Culture Collection, respectively. Oct4GiP cells were kindly provided by Dr. Austin Smith. ESCs were maintained on gelatin-coated plates in ESGRO complete plus clonal grade medium (Millipore, Billerica, MA). All transfections were performed as previously described (Hu et al, 2009; Zheng & Hu, 2012).
Direct RNA sequencing and data analysis
Direct RNA sequencing (DRS) was performed by Helicos BioSciences, and DRS reads were aligned to the mouse reference genome (mm9) using the index-DP genomic tool in Helisphere (Helicos BioSciences). Only uniquely mapped reads with a minimum mapped length of 25 and an alignment score of 4.0 were kept. We further filtered reads that arose from internal poly(A) priming, as previously described (Yao et al, 2012). For the remaining reads, we designated their 5′ ends as the corresponding poly(A) sites (PASs). To construct a consensus PAS annotation for downstream analysis, we clustered all individual PASs within a 40-nt window on the same chromosome strand and calculated a weighted coordinate as the designated PAS, as described previously (Yao et al, 2012). All DRS data have been deposited to NCBI SRA database (accession number: SRP025988).
All DRS read clusters were mapped to Ensemble genes. Next, we used Fisher's exact test to compare the ratio of the DRS read counts of one PAS to the sum of the read counts of all other PASs within the same gene. The P-values were adjusted by the Benjamini–Hochberg method to control false discovery rate (FDR). PASs with an FDR ≤ 10−4 were defined as significantly changed PASs. To create the scatterplot shown in Fig 2A, we selected two PASs with the smallest P-values for genes with multiple poly(A)s, and the PAS closer to the transcript start site is designated as the proximal PAS and the other as distal PAS. We then calculated the corresponding proximal/distal ratio. PAS pairs with an FDR ≤ 10−4 and |log10 (ES/Fip1 KD-proximal/distal ratio)| > 0.2 are highlighted. Genes with proximal-to-distal shifts are highlighted in blue, while those with distal-to-proximal shifts are highlighted in red.
In vitro cleavage/polyadenylation assay
PAS sequence (−100 nt to +100 nt of the cleavage site) of interest was cloned into pBluescript, and RNA substrate was synthesized by in vitro transcription in the presence of 32P-α-UTP. In vitro cleavage/polyadenylation was performed with HeLa nuclear extract under standard conditions, as previously described (Yao et al, 2012). For Fig 2D, nuclear extract was prepared from control HeLa cells and a HeLa cell line that stably expresses a Fip1-specific shRNA.
Mapping Fip1-RNA interactions by iCLIP-seq
Fip1 iCLIP-seq was carried out as previously described (Yao et al, 2012). For the analysis shown in Fig 5E, for all genes in each group, we first normalize the iCLIP signals by DRS read counts. For distal PAS, the DRS read count for the distal PAS was used. For proximal PAS, the total read count for both proximal and distal PAS was used as Fip1 iCLIP signals are from both the short and the long APA isoform. Then, we divided the cUTR or aUTR into 200 bins. Normalized iCLIP signals within the same bin from all genes in the same group were added up and then divided by the total number of genes within each group. The final normalized iCLIP signal (per gene) (y-axis) is plotted against the position within the 3′ UTR (percentage of UTR, x-axis). The iCLIP map in Supplementary Fig S25 (bottom panel) was made in the same way. For Supplementary Fig S25 (top panel), the iCLIP signals at the same position within the −100 nt to +100 nt region were added up and normalized as described above.
Acknowledgments
We thank Drs. M. Waterman, K. Hertel, K. Adelman, T. Hall, T. Kunkel, and P. Wade for critically reading of the manuscript; Drs. J. Ward and D. Fargo for assistance with bioinformatics analysis; Dr. C. Limoli for sharing equipment. This study was supported in part by the National Institute of Environmental Health Sciences, National Institutes of Health Intramural Research Program 1ZIAES102745-02 (to G. H., G. M. C., B. L., X. Z.), 1ZIAES102625-03 (to R. J., J. F.), NIH Grants GM090056 (to Y.S) and GM088342 (to Y.X.), American Cancer Society Grant RSG-12-186 (to Y.S), National Science Foundation Grant DBI-084621 (to X.X.), and a junior faculty grant from the Edward Mallinckrodt Jr. Foundation (to Y.X.).
Author contributions
GH and YS directed the project and wrote the manuscript with inputs from other authors. GH, YS, BL, YC, and GMC designed the experiments. BL, YC, GMC, XZ, and EAC executed most of the experiments. Bioinformatic analyses were performed by WL, XX, JW, YX, JF, PY, and RJ.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, Iori K, Rando TA. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell. 2012;10:327–336. doi: 10.1016/j.stem.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, Jaenisch R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Choi EA, Shi Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA. 2011;2:321–335. doi: 10.1002/wrna.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Miura RM, Tian B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics. 2006;22:2320–2325. doi: 10.1093/bioinformatics/btl394. [DOI] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011;43:853–866. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Paszkowski-Rogacz M, Nitzsche A, Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H, Hubner N, Doss MX, Sachinidis A, Hescheler J, Iacone R, Anastassiadis K, Stewart AF, Pisabarro MT, Caldarelli A, Poser I, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4:403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Elkon R, Drost J, van Haaften G, Jenal M, Schrier M, Oude Vrielink JA, Agami R. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol. 2012;13:R59. doi: 10.1186/gb-2012-13-7-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kuhn U, Menzies FM, Oude Vrielink JA, Bos AJ, Drost J, Rooijers K, Rubinsztein DC, Agami R. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012;149:538–553. doi: 10.1016/j.cell.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, Tian B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci USA. 2009;106:7028–7033. doi: 10.1073/pnas.0900028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Tian B. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE. 2009;4:e8419. doi: 10.1371/journal.pone.0008419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann I, Martin G, Friedlein A, Langen H, Keller W. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 2004;23:616–626. doi: 10.1038/sj.emboj.7600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lianoglou S, Garg V, Yang JL, Leslie CS, Mayr C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013;27:2380–2396. doi: 10.1101/gad.229328.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012;1:753–763. doi: 10.1016/j.celrep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AA, Cheung TH, Rando TA. All's well that ends well: alternative polyadenylation and its implications for stem cell biology. Curr Opin Cell Biol. 2013;25:222–232. doi: 10.1016/j.ceb.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Ng HH, Surani MA. The transcriptional and signalling networks of pluripotency. Nat Cell Biol. 2011;13:490–496. doi: 10.1038/ncb0511-490. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010;143:1018–1029. doi: 10.1016/j.cell.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi E, Lu J, Flanagan LA, Hertel KJ, Shi Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17:761–772. doi: 10.1261/rna.2581711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Alternative polyadenylation: new insights from global analyses. RNA. 2012;18:2105–2117. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, 3rd, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 2009;33:365–376. doi: 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol. 2001;17:435–462. doi: 10.1146/annurev.cellbio.17.1.435. [DOI] [PubMed] [Google Scholar]
- Takagaki Y, Seipelt RL, Peterson ML, Manley JL. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell. 1996;87:941–952. doi: 10.1016/s0092-8674(00)82000-0. [DOI] [PubMed] [Google Scholar]
- Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38:312–320. doi: 10.1016/j.tibs.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Oron E, Nelson B, Razis S, Ivanova N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell. 2012;10:440–454. doi: 10.1016/j.stem.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci USA. 2012;109:18773–18778. doi: 10.1073/pnas.1211101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature. 2002;416:545–548. doi: 10.1038/nature729. [DOI] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hu G. Oct4GiP reporter assay to study genes that regulate mouse embryonic stem cell maintenance and self-renewal. J Vis Exp. 2012;63:e3987. doi: 10.3791/3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.