

Abstract
RNA sequencing is widely used to measure gene expression across a vast range of animal and plant tissues and conditions. Most studies of computational methods for gene expression analysis use simulated data to evaluate the accuracy of these methods. These simulations typically include reads generated from known genes at varying levels of expression. Until now, simulations did not include reads from noisy transcripts, which might include erroneous transcription, erroneous splicing, and other processes that affect transcription in living cells. Here we examine the effects of realistic amounts of transcriptional noise on the ability of leading computational methods to assemble and quantify the genes and transcripts in an RNA sequencing experiment. We show that the inclusion of noise leads to systematic errors in the ability of these programs to measure expression, including systematic underestimates of transcript abundance levels and large increases in the number of false-positive genes and transcripts. Our results also suggest that alignment-free computational methods sometimes fail to detect transcripts expressed at relatively low levels.
Over the past decade, many computational methods have been developed to analyze data from RNA sequencing (RNA-seq) experiments (Li et al. 2010; Trapnell et al. 2012; Bray et al. 2016; Patro et al. 2017; Kovaka et al. 2019). The primary data from these experiments consist of a large collection of short sequencing reads, usually 100–150 bp in length, that themselves derive from transcribed RNA molecules in a tissue sample. Genome-guided transcriptome assembly methods (Trapnell et al. 2012; Maretty et al. 2014; Kovaka et al. 2019) map these reads to the genome of the target organism and then reconstruct and quantify full-length RNA molecules from the alignments. Alignment-free methods (Patro et al. 2014, 2017; Bray et al. 2016) use annotated transcripts to construct an index for exact lookup of short subsequences (k-mers). K-mers in each sequenced read are then matched against the index to determine which transcripts produced each read. These methods run much faster because they skip the alignment step, but they give up the ability to detect any genes or transcripts that are not already present in the annotation.
Both types of algorithms produce, for each transcript detected, an estimate of the level of expression of that transcript. These expression-level estimates are, in turn, used to determine expression values of full genes and to compute which genes and transcripts are differentially expressed in different experimental samples.
In testing and evaluating methods for RNA-seq data analysis, many published reports have relied on simulated data (Trapnell et al. 2012; Patro et al. 2014, 2017; Bray et al. 2016; Shao and Kingsford 2017; Kovaka et al. 2019). For example, Patro et al. (2017) used simulated data sets generated by Polyester (Frazee et al. 2015) and RSEM-sim (Li and Dewey 2011), Bray et al. (2016) used RSEM, and Kovaka et al. (2019) used FluxSimulator (Griebel et al. 2012) to generate reads. The reads themselves included sequencing errors, but all transcripts produced by the simulators were considered to be correct for the purposes of evaluating the RNA-seq abundance estimations. The need for simulations arises because we do not have an RNA-seq data set for which the ground truth is known, that is, for which we know precisely which genes and transcripts were expressed and at what levels they were present. Most evaluations therefore rely on a combination of real and simulated data to estimate the accuracy of these methods.
Several biases in RNA-seq protocols have been investigated as potential confounding factors in the downstream analysis (Patro et al. 2017; Ma and Kingsford 2019). These observations led to the development of targeted simulation protocols for accurate comparison of abundance quantification methods (Li and Dewey 2011; Frazee et al. 2015). Additionally, recent interest in the analysis of prespliced mRNA molecules (La Manno et al. 2018) led to modifications of quantification methods to account for the presence of unspliced isoforms for specific protocols (Bray et al. 2016).
Recent studies have shown that the human transcriptome has many “noisy” transcripts, that is, transcribed RNA sequences that do not represent functional genes (Struhl 2007; Cavallaro et al. 2020). These noisy transcripts have been estimated to comprise up to one-third of the RNA molecules in a cell, although most of them are present at very low levels (Van Bakel et al. 2010; Djebali et al. 2012; Palazzo and Lee 2015; Pertea et al. 2018). Despite this phenomenon, no previous study has simulated noisy transcripts as part of an evaluation of RNA-seq assembly or quantification programs. We wanted to test the hypothesis that the presence of noise might have a significant effect on the output of these programs.
In this study, we first developed a collection of methods to quantify the amount of noise in bulk RNA-seq experiments and to create simulated data sets that would reflect the types and abundance of transcriptional noise that were observed in large studies such as GTEx (The GTEx Consortium 2013). This noise includes intergenic transcripts, erroneous splicing, and incompletely spliced transcripts. We then generated a set of simulated RNA-seq experiments and analyzed them with three of the leading programs for estimating gene expression: StringTie2, Salmon, and kallisto.
Results
Properties of transcription in GTEx and simulation
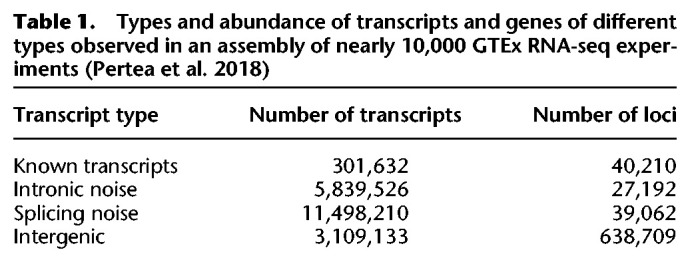
In this analysis, we investigated four distinct types of biological and technical variation, by partitioning transcriptome assemblies previously computed from the GTEx data set into (1) known transcripts, (2) erroneous transcripts caused by retained introns (“intronic noise”), (3) erroneous transcripts caused by the use of the wrong splice site (“splicing noise”), and (4) erroneous transcripts caused by transcription in intergenic regions (“intergenic”), as summarized in Table 1.
Table 1.
Types and abundance of transcripts and genes of different types observed in an assembly of nearly 10,000 GTEx RNA-seq experiments (Pertea et al. 2018)
In our analysis, we found that most known genes were expressed in at least one sample of a typical tissue (Fig. 1A). In contrast, fewer than half of both known loci and isoforms were actively expressed in a typical sample (Fig. 1B,C). We also found that known transcripts were more likely to occur in multiple samples of the same tissue (∼26%) compared with noisy transcripts (1.8% for intergenic noise, 0.5% for intronic noise, and 1.4% for splicing noise). Thus, although the complete GTEx data set contained a much higher number of noisy transcripts overall, at the level of a particular tissue, the number of noisy transcripts was generally lower than the number of real ones (Fig. 1B,C).
Figure 1.

Properties of the GTEx data set computed from transcriptome assemblies built for the CHESS database (Pertea et al. 2018) compared with simulated data. (A) Distributions of the number of annotated and intergenic loci observed per tissue. (B) Distributions of the number of annotated and intergenic loci observed per sample. (C) Distributions of the number of transcripts representing each noise type in a sample. (D) Fraction of expression in a typical sample that comes from real isoforms versus noisy isoforms. Only loci having both annotated and noisy transcripts being expressed are included. (E) Fraction of total expression from noisy transcripts in simulated samples.
Each type of transcription in our analysis displayed distinct expression properties within the data set. Importantly, we found known isoforms to dominate the expression within annotated genes. For a typical gene in our simulation, 80%–90% of the reads derived from known isoforms, similar to the proportion observed in the GTEx data set. As shown in Figure 1E, between 17% and 32% of transcription in our simulation comes from noisy isoforms (µ = ∼25%). This is comparable to the average of 25.7% noisy expression observed across samples in GTEx, with intergenic, intronic, and splicing noisy transcripts comprising on average 4.8%, 2.1%, and 18.8%, respectively, of the total expression (Supplemental Fig. S1).
We applied our simulation protocol (Methods) to create a data set composed of three tissues, each represented by 10 samples. Our comparisons between inter- and intra-sample properties of the simulated data set revealed that all targeted properties of the GTEx data set were preserved, while providing a high degree of randomization.
Abundance estimation
In our analysis below, we present results based on the cumulative contribution of different types of noisy expression to the output of RNA-seq analysis tools. A breakdown of the results by specific type of noise is presented in Supplemental Figures S2 through S8.
Transcript-level effects
For all methods considered here, the introduction of noisy expression led to a consistent increase in the number of transcripts falsely identified as expressed (Fig. 2A). False-positive rates (FPRs) and false-negative rates (FNRs) both in the absence and presence of noise are reported in Supplemental Figures S7 and S8. We observed that StringTie2 had both the smallest number of false positives (FPs) in the absence of noise (μ = 18,844; FPR = 7%) and the smallest increase in FPs, bringing its average up to 23,494 (∼25% increase; FPR = 8%). In comparison, Salmon had a slightly higher number of FPs in the absence of noise (μ = 21,546; FPR = 8%), but these had a much greater increase of ∼70% (μ = 36,677; FPR = 13%) in the presence of noise. The number of FP observations for kallisto was highest with noise-free data (μ = 34,316; FPR = 12%), and when noise was added, it produced the largest number of false-positive (FP) transcripts, averaging more than 51,000 (∼50% increase; FPR = 18%). On average, methods reported similar sets of FP transcripts across simulated samples with greater similarity observed between Salmon and kallisto (Supplemental Fig. S9).
Figure 2.

Effects of transcriptional noise on the transcript-level abundance estimation quantified across the 30 samples in the simulated data set. (A) Distribution of the number of false-positive (FP) observations per sample, with (brown) and without (blue) noise. (B) Expression levels assigned to FPs in the absence and presence of noise. (C) Distribution of the number of false-negative (FN) observations per sample. (D) Expression levels of FNs in the absence and presence of noise.
A common strategy to reduce FPs from RNA-seq analysis is to eliminate isoforms with low expression using predefined thresholds. To account for this in our analysis, we examined abundance estimates of the FP transcripts (Fig. 2B). We observed that StringTie2's FPs had the lowest median abundance, at 0.14 transcripts per million (TPM) with noise and 0.15 TPM without noise. Salmon and kallisto, in contrast, assigned substantially greater abundance to their FPs. Specifically, their median abundance estimates in the absence of noise were 0.4 TPM (Salmon) and 0.19 TPM (kallisto), and when noise was included, these increased to 0.85 and 0.39, respectively. In addition, Salmon and kallisto's abundance estimates showed a larger statistical dispersion, with many FPs having an expression level above 2.0.
If we used a minimum TPM threshold of one, as is sometimes done in RNA-seq analysis, then across 30 simulated samples in the absence of noise, Salmon and kallisto reported 262,085 and 290,537 FP transcripts, respectively, whereas StringTie2 reported 126,735. When noisy transcripts were added, all of these numbers went up, but the increases were much greater for Salmon and kallisto. In particular, across the 30 samples with noise, StringTie2 reported 171,087 FP transcripts with expression >1 TPM, whereas Salmon and kallisto reported 524,694 and 588,177, respectively.
We then evaluated the number of false negatives (FNs), that is, the number of transcripts that appeared in the simulated data but that each program failed to identify. For kallisto, we observed the smallest number of FNs in the absence of noise (μ = 1233; FNR = 5%) with an increase of ∼41% (FNR = 7%) after the introduction of noise (Fig. 2C). StringTie2 had more FNs (μ = 2109; FNR = 8%), but this number actually decreased by ∼1.1% when noise was added. We found that Salmon had the greatest number of FNs (μ = 3061; FNR = 12%), which increased ∼12% (FNR = 13%) after the introduction of noise. In contrast with the FPs, however, the FNs reported by StringTie2 were consistently different from those reported by Salmon and kallisto (Supplemental Fig. S9).
As with the FPs, StringTie2's FNs were expressed at very low levels, with nearly all of them having TPM < 1 (Fig. 2D). By looking at simulated abundance of FNs, we observed that both in the absence and presence of noise, StringTie2's FNs had a median expression of 0.4 TPM. The FNs for Salmon and kallisto, in contrast, had much higher median expression, at 2.02 and 1.84 TPM, respectively (for noise-free data), and slightly higher in the presence of noise. Additionally, among all transcripts with TPM > 1 across all 30 samples, Salmon and kallisto failed to identify 66,659 and 24,064 transcripts, respectively, compared with StringTie2 missing just 14,079 transcripts. When noise was introduced, this number increased to 14,289 for StringTie2, whereas Salmon's and kallisto's total FNs increased to 77,644 and 36,871, respectively.
We hypothesized that the introduction of transcriptional noise into the samples might increase abundance estimates proportionally with the number of noisy reads that overlapped annotated sequences. For all three methods, we observed reported abundances to be, on average, 20% lower than the estimate in the absence of noise (Supplemental Fig. S6). We suspect that the decrease occurs because noisy transcripts led the programs to report a greater number of FPs (as shown in Fig. 2A), which then absorbed many of the reads that instead should have been assigned to true-positive transcripts and altered the normalization factor of the TPM calculation.
Lastly, by analyzing the contributions of the three types of transcriptional noise, we found that >99% of the effects on RNA-seq analysis programs are caused by splicing noise (Supplemental Figs. S2–S8). Transcriptional noise that is entirely contained within introns or that is purely intergenic had little effect on the ability of any of the methods to accurately capture and quantify known annotated genes.
Gene-level effects
Transcriptional noise produces similar effects when we consider gene-level (as opposed to transcript-level) abundance estimates (Fig. 3; Supplemental Figs. S10, S11). A tradeoff between specificity and sensitivity was observed for all methods in our comparison: StringTie2 had the fewest FPs but the highest number of FNs, whereas Salmon and kallisto each had far more FPs but very few completely missed genes. The addition of transcriptional noise increased the number of FPs for all the methods (Fig. 3A), but in contrast to the transcript-level results, noise had almost no effect on the rate of FNs (Fig. 3B). Likewise, all three evaluated methods tended to report similar sets of genes as FPs on average, whereas overlaps between FNs were generally much smaller (Supplemental Fig. S12).
Figure 3.

Effects of noisy transcription on gene-level abundance estimation. (A) Distributions of the number of FP genes per sample, that is, the number of reported gene loci at which no actual transcripts were expressed. (B) Distributions of the number of FN genes per sample, that is, the number of gene loci for which the simulated data contained at least one expressed transcript but where the program failed to report any. (C) Percentage of change in the number of reads assigned to a gene as a function of the fraction of expression at that locus that comes from unannotated transcripts. Percentage of change was computed relative to the total number of reads simulated for all annotated transcripts at each locus. Only loci with more than zero reads from annotated transcripts are shown.
Furthermore, our analysis confirmed an expected relationship between the accuracy of expression estimates and the amount of noise relative to the expression of a locus (Fig. 3C): All methods were affected more in regions where more reads came from noise. We observed that the introduction of noise had a much greater effect on the accuracy of gene-level quantification of pseudoalignment algorithms, even though the estimates for loci where only a small fraction of expression came from noise were better than for alignment-based assembly methods.
Discussion
Our understanding of transcriptional processes in complex genomes is still incomplete. In particular, we do not yet know the extent of erroneous transcription, whether it is caused by splicing errors, read-through events, or other factors (Djebali et al. 2012; Palazzo and Lee 2015; Saudemont et al. 2017). Until the transcriptome is studied and understood more thoroughly, relying too strongly on a predefined set of expressed sequences may lead to substantial errors in the downstream analysis (Pickrell et al. 2010).
The experiments described here show that perfect, noise-free simulations present an inaccurate picture of the performance of methods for assembly and quantification of RNA-seq experiments. Although other biases in RNA-seq experiments have been shown to confound results (Li et al. 2010; Bray et al. 2016; Patro et al. 2017), the presence of transcriptional noise—that is, transcripts that do not represent functional genes—in the data may lead to both under- and overestimates of expression.
High numbers of FPs in expression analysis may propagate downstream in unexpected ways. Even in the absence of noise, our analysis showed that the leading programs generated thousands of FP transcripts, and the addition of noise added thousands more. These FPs, in turn, seemed to absorb many of the reads from true positives, with the result that all methods reduced their average estimates of expression levels by ∼20% when realistic amounts of transcriptional noise were present (Supplemental Fig. S6). Although we noticed a similar reduction of expression for all methods, the reasons for this change are probably different between methods. Although alignment-free methods incorrectly allocated reads to other annotated isoforms that were not expressed, StringTie2 used those reads to assemble novel isoforms, mostly at low expression levels.
After adding noisy transcription to our simulated data, we observed increases in both FPs and FNs as compared with noise-free controls. We speculate that such observations are primarily explained by the fact that the majority of loci in a given tissue express only a small number of functional molecules (Trapnell et al. 2010), while producing many overlapping nonfunctional splicing variants (Tress et al. 2017a,b). Reads from these noisy transcripts are sometimes counted toward expression of nonexpressed isoforms, creating FP results, such as the one illustrated in Supplemental Figure S13.
Repetitive elements in the genome could also explain some of the FPs that we observed. We found that close to 50% of bases in intronic and intergenic isoforms overlap regions masked by RepeatMasker (Smit et al. 2015), whereas splicing noise transcripts overlap a much lower proportion of repeats, similar to the fraction of repeats in known transcripts (Supplemental Fig. S14). However, because our analysis of the GTEx data set revealed that very few intronic and intergenic transcripts exist within a sample (Fig. 1C), the high percentage of repeats contained in those regions is unlikely to significantly alter the results.
In our analysis, we also observed a slight decrease in FN transcripts for StringTie2 after noise was added, in contrast with the increases observed for alignment-free methods. Because assembly methods are dependent on sufficient and complete coverage of sequenced molecules, the introduction of reads from noisy transcripts that overlap true positives may have helped cover some of the problematic areas, aiding in the assembly process.
At the gene level, StringTie2 had fewer than 1000 FPs, and this number increased only modestly when noise was present. In contrast, both Salmon and kallisto reported 6000–8000 FP genes, and these numbers increased by approximately 5000 in the presence of noise. However, the number of completely missed genes (FNs) for both Salmon and kallisto was very low, regardless of the presence of noise. This makes sense as both programs rely heavily on a predefined gene list that, in our simulations, contained all the true-positive genes. Thus, they were able to detect the true genes accurately, even those expressed at low levels.
Another phenomenon observed here was that when abundance was measured in TPM, transcriptome assembly methods such as StringTie2 inherently produced lower abundance estimates than did alignment-free methods, because TPMs are normalized with respect to the effective length of transcripts. Annotation-dependent methods will always have the same total length of expressed transcripts, which is provided by the reference annotation. In contrast, assembly methods produce a unique transcriptome for each experiment, which affects the TPM normalization for length and results in lower TPM values when the fragmentation of the transcriptome is increased. This finding is in agreement with previous reports (Zhang et al. 2017), and although this phenomenon tends to result in an underestimate of expression values, the same property may aid in the filtering of FPs.
We should point out that some of the transcripts that were considered noisy in the CHESS data set, and from which we modeled our simulated noisy data, might in fact come from rarely expressed but functional isoforms. The results presented here were computed relative to the set of annotated transcripts, and therefore, the conclusions about how their abundances are affected should not change if some of those isoforms later turn out to be real.
Although our findings indicate that all methods are challenged by the presence of transcriptional noise, effects on accuracy differ among the methods. For applications that require higher specificity, Salmon and kallisto might be preferred (Figs. 2, 3). With 90% of total expression coming from real isoforms in a typical gene (Fig. 1D), all methods showed highly accurate abundance estimation (Fig. 3C). Similar observations extend to the transcript level. However, in applications in which one is interested in knowing the precise isoform mixture at each locus, a lower rate of FPs might be preferred, in which case StringTie2 has an edge because of its ability to assemble unannotated isoforms and because its FPs have lower abundances.
It is important to understand that our analysis might in fact be underestimating the scope of the problem. Our filtering criteria, which we used to remove redundancy and less prevalent types of noisy transcription from GTEx, resulted in the removal of approximately 10 million assembled molecules, nearly one-third of the total number. Because nonintergenic noisy transcripts not included in our analysis would by definition have overlapped annotated features, the bias introduced by the reads contained in those transcripts would likely lead to a greater effect on the accuracy of RNA-seq analysis programs.
Finally, we hope that the approach used in this study will guide future assessments of RNA-seq abundance quantification methods by providing a set of simulated data sets that were based on curated experimental data and that include realistic amounts of transcriptional noise outside of the annotated transcripts. If new tools and protocols continue to be evaluated without accounting for unannotated transcription, we will be left with an incomplete and possibly erroneous perception of their performance.
Methods
The tools described here were designed to create realistic simulations at the multitissue level by computing parameters from nearly 10,000 RNA-seq experiments produced as part of the GTEx project (The GTEx Consortium 2013). We computed gene- and transcript-level expression values from the simulated data using three state-of-the art quantification methods: StringTie v.2.1.2, Salmon v.0.14.0, and kallisto v.0.46.1.
Data filtering
We examined transcriptome assemblies of the GTEx data set from the CHESS project (Pertea et al. 2018) at the level of individual samples, at the level of individual tissues, and across the full data set. To reduce the number of confounding factors in our analysis, we removed any noisy isoforms that overlapped annotated loci on the opposite strand, contained annotated loci within their introns, or were in close proximity of known genes but did not overlap their exons. After filtering, we retained 20,748,278 assembled isoforms out of the initial set of about 30 million.
Typing of transcripts
Transcripts were compared to the full database of CHESS genes and transcripts using gffcompare (Pertea and Pertea 2020). We labeled a transcript as real if all its introns matched a transcript found in the CHESS annotation (small differences in the positions at the beginning and end of transcription were disregarded). If an isoform was contained within an intronic region of a known gene, it was labeled as intronic noise. Splicing variants that overlapped known loci but that contained unannotated exons, introns, or exon chains were labeled as splicing noise. Transcripts sharing no overlap with the annotated loci were labeled as intergenic noise (Table 1).
Quantification
By using the mappings between annotated and noisy transcripts across levels of assembly, we quantified the following parameters for the GTEx data set:
The number of real and intergenic loci expressed in each tissue and sample.
The number of transcripts of each type and their corresponding TPM values. For each locus (gene), observations from all samples per each tissue were grouped together.
The number of reads in each sample.
Simulated tissue and sample generation
After choosing a set of transcripts from randomly selected loci to be expressed in a tissue, we proceeded by generating a set of possible expression values for each transcript. In this step, observations from different types of transcripts (real + noisy) were treated jointly for each locus in a simulated sample. Sample-level observations were similarly grouped and treated jointly at the tissue level. This step was required to preserve the inherent relationships between (1) transcripts of different types in a single sample and (2) expression of the same transcript in different samples from the same tissue.
Annotations and expression values for each sample were simulated next by randomly picking one set of possible transcript observations for each locus. The order of transcripts and corresponding expression values were shuffled before being linked and remained constant for each sample. This guaranteed preservation of any relationships between expressions of transcripts in different samples of the same tissue, observed in the modeled data set (Fig. 1A).
Read counts per transcript
We then calculated the expression values to be used with the Polyester simulator. Polyester requires coverage to be provided for each transcript in a simulation (Frazee et al. 2015). To compute the target number of reads to be simulated, we reversed the TPM calculation:
where Ci is the coverage of transcript i, Ei is its expression measured in TPM, Li is length, N is the number of reads in the sample, and l is the read length (Li and Dewey 2011).
Simulation parameters
For our analysis, we simulated three hypothetical tissues, each containing 10 samples. Single-end 101-bp reads for each sample were generated using Polyester with an error rate of 0.4%.
In our preliminary analysis, we noticed Polyester was unable to accurately model paired-end sequences. In particular, for transcripts shorter than the fragment length, Polyester left gaps in the coverage. We also observed that Polyester was unable to extend read coverage to the end of the last exon in the transcript when simulating single-end reads. We were able to bypass these issues by simulating single-end reads with Polyester and setting the fragment length to be the same as the read length, with a standard deviation of zero. We combined reads generated from real transcripts, splicing noise, intronic noise, and intergenic noise transcripts together for each sample.
Analysis
For quantification of genes and transcripts, we used three of the most widely used current methods for transcriptome quantification: StringTie2, Salmon, and kallisto. Each method was run using the recommended parameters, as described in the Supplemental Methods. For StringTie2, alignments were produced using HISAT2 v.2.2 (Kim et al. 2019).
To avoid unnecessary complexity, we restricted our analysis to the primary chromosomes of the GRCh38.12 human assembly (Schneider et al. 2017), excluding all alternative scaffolds and patches. For annotation, we used the version of CHESS 2.2 human gene catalog tailored for transcriptome assembly, which is also restricted to main scaffolds only.
Additionally, in our analysis we took care to avoid creating differences in gene expression that might be owing to the normalization method. TPM is widely used to measure gene expression because it is more stable than other abundance metrics (Conesa et al. 2016); however, they are dependent on the cumulative effective length of the underlying transcriptome being quantified. Because our comparison includes a method (StringTie2) that discovers novel isoforms, the normalization factor in TPM computation is very different from the one used by Salmon and kallisto, which rely exclusively on a predefined annotation. These differences result in different TPM values, even where the read counts and inferred transcript lengths are the same.
Wherever possible, therefore, normalized expression values were compared as a percentage of change from estimates obtained in the absence of noise to estimates computed in the presence of noise within each method. For fairness when evaluating FNs, simulated TPMs were computed based on all transcripts present in a sample (real, splicing, intronic, intergenic) as well as all transcripts in the sample that matched the annotation.
Gene-level abundance
Each method in our analysis estimates abundances at the transcript level by default. Computing abundances at the gene level involves using separate tools for different methods. Abundance measurements such as TPM typically factor cumulative effective length of the transcribed sequences into the equation (Li and Dewey 2011). This presents a distinct challenge for comparing annotation-agnostic methods such as StringTie2 to pseudomapping approaches like Salmon and kallisto, which always rely on a predefined set of transcribed sequences. To reduce the impact of the difference in normalization factors, we performed gene-level abundance comparisons based on the raw read-count aggregation.
Software availability
Our simulation and evaluation protocols are available as Supplemental Code as well as from GitHub (https://github.com/alevar/simann, and https://github.com/alevar/tx_noise).
Data access
All assemblies, mapping files, filtered data, and simulated data from this study are provided at CyVerse Data Commons (https://doi.org/10.25739/v903-wd86) and ftp://ftp.ccb.jhu.edu/pub/avaraby/RNAseqNoise.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This work was supported in part by grant DBI-1759518 from the National Science Foundation and grant R01-HG006677 from the U.S. National Institutes of Health. We thank Dr. Martin Steinegger, Dr. Christopher Pockrandt, and Dr. Jennifer Lu for the helpful discussions during the method's development and analysis.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.266213.120.
References
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Cavallaro M, Walsh MD, Jones M, Teahan J, Tiberi S, Finkenstädt B, Hebenstreit D. 2020. 3′-5′ crosstalk contributes to transcriptional bursting. bioRxiv 10.1101/514174 [DOI] [PMC free article] [PubMed]
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A. 2016. A survey of best practices for RNA-seq data analysis. Genome Biol 17: 13 10.1186/s13059-016-0881-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazee AC, Jaffe AE, Langmead B, Leek JT. 2015. Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics 31: 2778–2784. 10.1093/bioinformatics/btv272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel T, Zacher B, Ribeca P, Raineri E, Lacroix V, Guigó R, Sammeth M. 2012. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res 40: 10073–10083. 10.1093/nar/gks666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. 2019. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20: 278 10.1186/s13059-019-1910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. 2018. RNA velocity of single cells. Nature 560: 494–498. 10.1038/s41586-018-0414-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. 2010. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26: 493–500. 10.1093/bioinformatics/btp692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Kingsford C. 2019. Detecting, categorizing, and correcting coverage anomalies of RNA-seq quantification. Cell Syst 9: 589–599.e7. 10.1016/j.cels.2019.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretty L, Sibbesen JA, Krogh A. 2014. Bayesian transcriptome assembly. Genome Biol 15: 501 10.1186/s13059-014-0501-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, Lee ES. 2015. Non-coding RNA: what is functional and what is junk? Front Genet 6: 2 10.3389/fgene.2015.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Mount SM, Kingsford C. 2014. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol 32: 462–464. 10.1038/nbt.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G, Pertea M. 2020. GFF utilities: GffRead and GffCompare. F1000Res 9: 304 10.12688/f1000research.23297.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Shumate A, Pertea G, Varabyou A, Breitwieser FP, Chang YC, Madugundu AK, Pandey A, Salzberg SL. 2018. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol 19: 208 10.1186/s13059-018-1590-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, Pritchard JK. 2010. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet 6: e1001236 10.1371/journal.pgen.1001236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudemont B, Popa A, Parmley JL, Rocher V, Blugeon C, Necsulea A, Meyer E, Duret L. 2017. The fitness cost of mis-splicing is the main determinant of alternative splicing patterns. Genome Biol 18: 208 10.1186/s13059-017-1344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864. 10.1101/gr.213611.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Kingsford C. 2017. Accurate assembly of transcripts through phase-preserving graph decomposition. Nat Biotechnol 35: 1167–1169. 10.1038/nbt.4020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2015. RepeatMasker Open-4.0. 2013–2015. http://www.repeatmasker.org.
- Struhl K. 2007. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol 14: 103–105. 10.1038/nsmb0207-103 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress ML, Abascal F, Valencia A. 2017a. Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci 42: 98–110. 10.1016/j.tibs.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress ML, Abascal F, Valencia A. 2017b. Most alternative isoforms are not functionally important. Trends Biochem Sci 42: 408–410. 10.1016/j.tibs.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bakel H, Nislow C, Blencowe BJ, Hughes TR. 2010. Most “dark matter” transcripts are associated with known genes. PLoS Biol 8: e1000371 10.1371/journal.pbio.1000371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang B, Lin LL, Zhao S. 2017. Evaluation and comparison of computational tools for RNA-seq isoform quantification. BMC Genomics 18: 583 10.1186/s12864-017-4002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.