

Abstract
Transgenic T-cell receptor (TCR) T cell–based adoptive cell therapies for solid tumors are associated with dramatic initial response rates, but there remain many instances of treatment failure and disease relapse. The association of infusion product cytokine profiles with clinical response has not been explored in the context of TCR T-cell therapy products. Single-cell antigen-dependent secretomic and proteomic analysis of preinfusion clinical TCR T-cell therapy products revealed that TNF-α cytokine functionality of CD8+ T cells and phospho-STAT3 signaling in these cells were both associated with superior clinical responsiveness to therapy. In contrast, CD4+ T-helper 2 (Th2) cell cytokine profiles were associated with inferior clinical responses. In parallel, preinfusion levels of IL-15, Flt3-L, and CX3CL1 were all found to be associated with clinical response to therapy. These results have implications for the development of therapeutic biomarkers and identify potential targets for enrichment in the design of transgenic TCR T-cell therapies for solid tumors.
Keywords: Cellular Immunotherapies, cytokines, soft-tissue sarcomas, melanoma, single-cell technologies
INTRODUCTION
Cancer immunotherapy with T cells expressing a transgenic T-cell receptor (TCR-T) or chimeric antigen receptor (CAR) can generate objective clinical responses in a wide variety of solid tumors. However, while these treatments lead to dramatic initial clinical responses in some patients, there are many patients who do not respond at all to the initial infusion of transgenic T cells, and the responses to therapy that do occur are often not durable beyond 6-12 months (1,2). There is substantial heterogeneity in the biological activities of the individual cells infused due to functional differences resulting from the specific secretomes/proteomes that are unique to each single cell. Given the high degree of variability seen in cell populations within a given setting, analysis of single cells is necessary for uncovering mechanisms that cannot be identified on a bulk population level. Previous work with CD19-targeted CAR T cells has demonstrated that the cytokine polyfunctionality of the cell therapy infusion product is associated with superior clinical response rates (3). Furthermore, the levels of certain cytokines (e.g. IL-7 and IL-15) within the patients’ serum at the time of cell therapy infusion (owing to the effectiveness of lymphodepleting chemotherapy regimens) have also been associated with clinical response to therapy with both CAR T cells and autologous tumor-infiltrating lymphocytes (3,4). However, neither of these phenomena have been explored in the setting of transgenic TCR T cell therapy for solid tumors.
Given the importance of this knowledge gap, we explored these factors in the final infusion cell therapy products and preinfusion sera of patients with melanoma and sarcoma positive for MART-1 and NY-ESO-1 treated with TCR T-cell therapies targeting MART-1 and NY-ESO-1 (1,2). We hypothesized that the therapeutic activity of the TCR T cells is mediated via their specific cytokine secretion programs working in tandem with the cytokine milieu of the host following lymphodepleting chemotherapy conditioning (prior to the cell product infusion); the latter can play an important role in the functionality of the infused TCR T cells. Herein, we describe an exploratory observational analysis of single-cell multiplexed cytokine polyfunctionality (5), single-cell phospho-protein signaling, and preinfusion sera in a cohort of patients treated with transgenic TCR T-cell therapies, and their associations with clinical responsiveness and antitumor activity. We report that products containing cytokine functionalities characterized by high CD8+ T cell TNFα activity, low CD4+ T-helper 2 (Th2) cell activity, and high CD8+ T cell STAT3 activity are associated with superior responsiveness to therapy.
METHODS
Clinical trial, patients, and manufacturing of MART-1 and NY-ESO-1 TCR T cells
For the F5-MART-1 transgenic TCR adoptive cell therapy clinical trial, 15 patients positive for HLA-A*0201 and with MART-1+ metastatic melanoma were enrolled under NCT00910650 (UCLA IRB #08-02020 and #10-001212) from April 2009 to September 2011, under investigational new drug (IND) #13859 (1). For the NYESO-1 transgenic TCR adoptive cell therapy clinical trials, 13 patients positive for HLA-A*0201 and with NYESO-1-+ sarcoma or melanoma were enrolled under NCT02070406 or NCT01697527 (UCLA IRB #12-000153 and #13-001624, respectively) under IND#15167 (2), or NCT03240861 (UCLA IRB #15-000511) under IND#15167 and IND#17471. Written consent was obtained for each patient, and all studies were conducted in accordance with local regulations, the guidelines for Good Clinical Practice, and the principles of the Declaration of Helsinki. Clinical trial design, manufacturing of the MART-1 and NYESO-1 TCR-T cells, and expansion/contraction kinetics of TCR+ transgenic T cells in vivo have been previously described (1,2). Briefly, non-mobilized autologous PBMCs were stimulated in culture with IL2 (Clinigen NDC 76310-022-01) and OKT3 (Miltenyi Biotec 170-076-124) and transduced with clinical grade MSCV retrovirus vector expressing the MART-1 F5 TCR or the NYESO-1 TCR on two consecutive days, then continually expanded ex vivo for 6-7 days. Up to 1x109 transgenic TCR T cells were administered to each patient following conditioning chemotherapy with cyclophosphamide and fludarabine, along with post-infusion systemic IL2 for 7-14 days, and dendritic cell vaccine boosts, as previously described (1,2). For patients treated per NCT03240861, up to 1x109 transgenic TCR T cells were administered to each patient following conditioning chemotherapy with busulfan and fludarabine, along with post-infusion systemic IL-2 for 7 days and autologous stem cell rescue with minimum 2.5x106/kg lentiviral transduced TCR hematopoietic stem cells. PBMC samples from the infusion product, as well as from serial peripheral blood samples after adoptive cell transfer, were collected as previously described (1,2) and stored in liquid nitrogen prior to analysis. Patients’ responses to therapy were determined per RECIST1.1 criteria (6), these responses, progression free survival, overall survival, and TCR kinetics in vivo have been previously published (1,2).
Cell lines
HLA-A*02:01-transfected K562 cells (7) were a gift from Wolfgang Herr in 2003. Cells were authenticated annually via flow cytometry against HLA-A*02:01 (BD Biosciences 558570). Cells were cultured in RPMI1640 media (Gibco 11875–093) supplemented with 10% FBS (Sigma F2442), 1xGlutamax (Gibco 35050061), 1xPenecillin/Streptomycin (Thermo 15140122), hereafter referred to as “complete media,” and passaged every 3-4 days. Cells were passaged 2-3 times until used in our assays before being discarded. Cells were also supplemented with 1μg/mL Geneticin (ThermoFisher 10131027) until subsequent co-culture experiments described below. All cell cultures were tested for Mycoplasma contamination and found negative via the MycoAlert Kit (LT07–318).
Analysis of single-cell cytokine secretion
Infusion product samples from clinical transgenic MART-1 or NY-ESO-1 TCR-T cell products (n = 27) were cultured at a density of 106 cells/mL in complete media and 20ng/mL recombinant human IL2 (BioLegend 589104) for overnight recovery. CD4+ and CD8+ T-cell subsets were then separated using anti-CD4 and anti-CD8 microbeads (Miltenyi Biotec, 130-045-201 and 130-045-101), respectively, and stimulated with K562 cells transduced with HLA-A:02:01 either alone or pulsed with (1 μg/μL) at a final concentration of 12 μg/mL corresponding target peptide (MART-126–35, ELAGIGILTV, or NY-ESO-1157–165, SLLMWITQC), representing tonic and stimulated conditions, respectively, at an effector:target ratio of 1:1 for 24 hours at 37°C, 5% CO2. This allowed for the MHC I–restricted transgenic TCR, which was expressed on CD8+ and CD4+ T cells alike, to be stimulated. The cocultured CD4+ and CD8+ T cells were then further enriched using anti-CD235a–conjugated magnetic beads (PhenomEx, DEPLETION-1001-1) to deplete the K562 cells. CD4+ and CD8+ TCR T cells were stained with anti-CD4 or anti-CD8 conjugated to Alexa Fluor 647 (PhenomEx STAIN-1002-1 and STAIN-1003-1) at room temperature for 10 minutes, rinsing once with PBS, and resuspending in complete media at a density of 1x106/mL. Approximately 30 μL of the cell suspension was loaded onto the PhenomEx single-cell barcode chip (SCBC, PhenomEx, ISOCODE-1001-04) for single-cell secretomic evaluation or bulk acellular cytokine quantification.
Protein secretions from ~1,000 single cells were captured by the 32-plex antibody barcoded SCBC and analyzed by fluorescence ELISA–based assay on an IsoSpark platform for 20 hours under controlled temperature and CO2 conditions following single-cell isolation within the microfluidic chip, as previously described (8). Cytokines interrogated were granzyme B, perforin, IFNγ, MIP-1α, TNFα, TNFβ, GM-CSF, IL2, IL5, IL7, IL8, IL9, IL12, IL15, IL21, IL4, IL10, IL13, IL22, TGFβ, sCD137, sCD40L, CCL11, IP-10, MIP-1β, RANTES, IL1β, IL6, IL17A, IL17F, MCP-1, MCP-4. Polyfunctional TCR T cells (secreting 2 or more cytokines per cell) were evaluated using IsoSpeak software v1.11.0 (PhenomEx). Assay quality was determined using metrics that included cell counts, low background noise, and analyte detection above thresholds, as specified by the manufacturer. All data were normalized by viability and transduction. The Polyfunctional Strength Index (PSI) of CD4+ and CD8+ TCR T cells was computed using a prespecified formula, defined as the percentage of polyfunctional cells, multiplied by signal intensity of the proteins secreted by those cells, as previously described (8). The Functional Strength Index (FSI) of CD4+ and CD8+ TCR T cells was similarly calculated, defined as the percentage of cells secreting a given cytokine, multiplied by signal intensity of the protein secreted, as previously described (5). Overall PSIs (i.e. average of the total CD4+ and CD8+ PSIs) were also calculated, as previously described (3). CD4+ Th cell cytokine polyfunctionality profiles were defined as Th1 (IFNγ, IL2), Th2, (IL4, IL5, IL10, and IL13), Th9 (IL9), or Th17 (IL17A, IL17F, and IL22).
Acellular cytokine analysis
Co-culture media from the antigen stimulation conditions described above was frozen until time for analysis, at which point it was thawed and loaded into CodePlex chips (PhenomEx) according to the manufacturers’ instructions. Samples were analyzed in replicate (5μL per replicate) with the same 32-plex antibody panel as above on an IsoSpark platform (PhenomEx) and calibrated to internal standards. Outputs were averaged between replicates and quantified in pg/mL.
Analysis of single-cell intracellular phospho-proteome and intracellular cytokines
Infusion product samples from clinical transgenic NY-ESO-1 TCR T-cell products (n = 13) were recovered for 24 hours in RPMI with 20 ng/mL IL2 (BioLegend). After recovery, CD4+ and CD8+ T cells were enriched using anti-CD4 and anti-CD8 microbeads (Miltenyi) as above. If viability was below 50% by trypan blue exclusion, cells were centrifuged through Ficoll Paque Plus (GE Healthcare 17-1440-03) mixed with complete RPMI. Viable cells were isolated within the Ficoll–media interface and then resuspended in complete RPMI media prior to enrichment. After enrichment, CD8+ and CD4+ cells were resuspended at 2 x 106 cells/mL in complete RPMI with IL2 for 20 hours. CD4+ and CD8+ T cells were then immediately cocultured with either NY-ESO-1 peptide–pulsed or non-peptide–pulsed K562 cells in a 96 well U bottom plate at a ratio of 1:1 for 18 hours, representing stimulated and tonic signaling conditions, respectively. Brefeldin A (ThermoFisher/Invitrogen 00-4506-51) was added to each well (1:1000 dilution) and cells were incubated for an additional 2 hours. Following coculture, K562 cells were depleted using anti-CD235a beads (Invitrogen/ThermoFisher Scientific). After depletion, the cells were resuspended in complete RPMI without IL2 and stained with anti-CD4 or anti-CD8 conjugated to Alexa Fluor 647 (PhenomeX) as above for on-chip cell detection. Stained cells for each condition were resuspended in PBS to a cell density of 1.25 x 106 cells/mL supplemented with 1:100 protease phosphatase inhibitor cocktail (Cell Signaling Technology 5872) and 1:200 Calyculin A (Cell Signaling Technology 9902S). Each Human Adaptive Immune Signaling Chip (PhenomeX, ISOCODE-4L04-4-C), patterned with an antibody barcode against 15 intracellular cytokines and phosphoproteins, was loaded with 36 μL (~45,000 cells). Intracellular cytokines interrogated were GM-CSF, granzyme B, IFNγ, IL10, IL2, IL8, MIP-1β, perforin, and TNFα, and phosphoproteins interrogated were p-IκBα, p-MEK1-2, p-NF-κB p65, p-STAT1, p-STAT3, and p-STAT5. Chips were loaded onto an IsoLight, which processes IsoPlexis/PhenomeX chips using automated incubation, fluidics, and imaging. Cells were immediately lysed following individual cell isolation in the microfluidic chip prior to antibody incubation and fluorescence imaging/quantifiation. IsoSpeak software was used to analyze the data from both the stimulated and tonic culture conditions. FSI and single secretor data were generated with internal assistance in IsoSpeak. Assay quality was determined using metrics that included cell counts, low background noise, and analyte detection above thresholds, as specified by the manufacturer. All data were normalized by viability and transduction.
Serum cytokine analysis
Serum cytokine levels were assessed in serum samples taken from patients on the day of transgenic TCR T cell infusion (but prior to the infusion itself). Cytokine levels were assessed using the Luminex xMAP® Immunoassay, which was performed in the UCLA Immune Assessment Core. The human 38-plex magnetic cytokine/chemokine (EMD Millipore) panel was used according to the manufacturer’s instructions. This allowed for quantification of EGF, FGF-2, eotaxin, TGFα, G-CSF, Flt-3L, GM-CSF, CX3CL1 (fractalkine), IFNα2, IFNγ, GRO, IL10, MCP-3, IL12P40, MDC, IL12P70, IL13, IL15, sCD40L, IL17A, IL1Rα, IL1α, IL9, IL1b, IL2, IL3, IL4, IL5, IL6, IL7, IL8, IP-10, MCP-1, MIP-1α, MIP-1β, TNFα, TNFβ, and VEGF. Briefly, 25μL undiluted plasma samples were mixed with 25 μL magnetic beads, and allowed to incubate overnight at 4°C while shaking. After washing the plates twice with wash buffer in a Biotek ELx405 washer, 25 μL of biotinylated detection antibody was added and incubated for 1 hour at room temperature. 25 μL streptavidin-phycoerythrin conjugate was then added to the reaction mixture and incubated for another 30 minutes at room temperature. Following two washes, beads were resuspended in sheath fluid, and fluorescence was quantified using a Luminex 200TM instrument.
MHC dextramer immunologic assessment of surface expression of transgenic TCRs
Analysis using fluorescent HLA-A:02:01 MHC dextramers with corresponding MART-1 and NY-ESO-1 peptides (Immudex, WB2162 and WB2696), which enable detection and quantification of corresponding MART-1 and NY-ESO-1 TCRs (Immudex) was performed on patient infusion products and post-infusion ex vivo samples, as previously described (1,2,9). Our definitions for a positive or negative immunologic response using standardized MHC multimer assays were used, which are based on assay performance specifications by defining changes beyond the assay variability with a 95% confidence level (9).
Statistical analysis
Graphing and statistical analyses were performed using GraphPad Prism V.9 (GraphPad RRID:SCR_002798). Comparisons between PSI/FSI values of responder vs non-responder cohorts utilized the unpaired t-test, while correlations between PSI/FSI values and progression free survival, overall survival, peak TCR% in circulation, or peak percent change in tumor burden, using the sum of the diameters (longest for non-nodal lesions, short axis for nodal lesions) relative to baseline measurements, per RECIST1.1 criteria (6), utilized Pearson’s correlation (r). p values of <0.05 were considered significant for all analyses.
Data Availability
The data generated in this study are available within the article and its Supplementary Data Files or upon request from the corresponding author.
RESULTS
Trial Conduct, Patient Characteristics, and Outcomes
27 patients from our previous trials evaluating adoptive cell therapy with TCR T cells directed against MART-1 and NY-ESO-1 (1,2) were selected for analysis. Patient demographics, clinical characteristics, and outcomes are summarized in Table 1. Following conditioning chemotherapy, patients were treated with up to 1 × 109 autologous transgenic TCR T cells. Of the total patients selected, 17 of 27 patients treated demonstrated a transient objective response to therapy, defined as a reduction in measurable tumor burden compared to baseline/pretreatment values, per RECIST1.1 criteria.
Table 1.
Demographics and outcomes for patients receiving MART-1 or NY-ESO-1 TCR-T cell therapy at UCLA.
| Patient study number | Transgenic TCR target | Sex (M/F) | Age | Type of Cancer | Active Disease Sites | Evidence of transient tumor response via PET/CT | Response at EOS (day 90) | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|---|---|---|---|
| F5-1 | MART-1 | M | 60 | Melanoma | Lung, Stomach, Liver, Pancreas, Peritoneum, SC | Yes | PD | 3 | 5 |
| F5-2 | MART-1 | F | 46 | Melanoma | Skin, LN, bone | Yes | SD | 6 | 10 |
| F5-3 | MART-1 | M | 61 | Melanoma | Lung, Liver | Yes | SD | 7 | 86 |
| F5-4 | MART-1 | M | 50 | Melanoma | Lung, LN, SC | No | PD | 2 | 22 |
| F5-6 | MART-1 | M | 59 | Melanoma | Lung, LN | No | SD | 3 | 4 |
| F5-7 | MART-1 | M | 48 | Melanoma | SC, Bone | Yes | SD | 4 | 11 |
| F5-8 | MART-1 | M | 44 | Melanoma | LN, Liver | Yes | SD | 4 | 11 |
| F5-9 | MART-1 | F | 46 | Melanoma | Skin, LN | No | PD | 3 | 20 |
| F5-10 | MART-1 | F | 47 | Melanoma | Lung, adrenal, LN, SC, orbit | Yes | PD | 2 | 8 |
| F5-11 | MART-1 | F | 56 | Melanoma | Lung, LN | Yes | SD | 4 | 4 |
| F5-12 | MART-1 | M | 40 | Melanoma | Lung, LN | Yes | SD | 5 | 8 |
| F5-13 | MART-1 | M | 60 | Melanoma | Lung, Abdomen, SC | Yes | SD | 3 | 8 |
| F5-14 | MART-1 | F | 50 | Melanoma | Lung, abdomen, SC | No | PD | 0.5 | 1 |
| F5-15 | MART-1 | M | 76 | Melanoma | Right axilla, lung, chest wall, liver | No | PD | NA | 0.5 |
| ESO-1 | NY-ESO-1 | M | 47 | Liposarcoma | Right Renal Fossa, Liver, Peritoneal | No | PD | 2.6 | 16 |
| ESO-2 | NY-ESO-1 | M | 37 | Malignant Peripheral Nerve Sheath Tumor | Right iliopsoas, peritoneal, retroperitoneal | No | NA | NA | 1.3 |
| ESO-3 | NY-ESO-1 | F | 24 | Synovial Sarcoma | Lung | Yes | PR | 96 | 96 |
| ESO-4 | NY-ESO-1 | M | 41 | Synovial Sarcoma | Left Infraclavicular Mass; Left Pectoralis Mass | Yes | PD | 3 | 25 |
| ESO-5 | NY-ESO-1 | F | 43 | Synovial Sarcoma | Right popliteal fossa; Lung | Yes | PR | 9 | 41.5 |
| ESO-6 | NY-ESO-1 | M | 26 | Osteosarcoma | Lung | Yes | PD | 2.5 | 19 |
| INY-1 | NY-ESO-1 | M | 51 | Synovial Sarcoma | Lung, bone marrow | No | PD | 2 | 2 |
| INY-2 | NY-ESO-1 | M | 66 | Melanoma | LN, Liver | Yes | PD | 3 | 6 |
| INY-3 | NY-ESO-1 | F | 44 | Synovial Sarcoma | Right popliteal fossa; Lung | Yes | PD | 3 | 31 |
| INY-4 | NY-ESO-1 | M | 24 | Melanoma | Lung, LN, adrenal gland, liver, trachea, brain | No | PD | NA | 3 |
| NYSCT-01 | NY-ESO-1 | M | 27 | Epithelioid Sarcoma | Right forearm, humerus, lung | No | PD | 2 | 19.5 |
| NYSCT-03 | NY-ESO-1 | F | 28 | Synovial Sarcoma | Right chest wall, lung | Yes | PR | 3 | 3.5 |
| NYSCT-05 | NY-ESO-1 | M | 39 | Synovial Sarcoma | Lung | Yes | PD | 3 | 9 |
EOS – end-of-study, SD - stable disease, PR - partial response, PD - progressive disease, LN – lymph node, SC – subcutaneous
CD8+ T-cell TNF-α and STAT3 activity are associated with superior clinical response to TCR T cell therapy
To evaluate the cytokine functionality of the cell therapy infusion products, we performed single-cell cytokine secretion assays on each patient’s infusion product following co-culture of the cells with HLA-A:02:01-expressing K562 cells pulsed with the corresponding target antigen peptide (MART-1 or NY-ESO-1). We observed that the CD8+ T-cell TNFα FSI was significantly greater in infusion product cells given to patients with a measurable clinical response to therapy (responders) as opposed to cells given to patients without a clinical response (non-responders) (Figure 1A). Additionally, we found that the CD8+ T-cell TNFα FSI correlated with superior patient survival (Figure 1B), while the overall (average between CD4+ and CD8+ T cells) TNFα PSI, i.e. the percentage of single-cells secreting two or more cytokines multiplied by the degree of cytokine secretion intensity, correlated with the degree of antitumor response, as measured by peak percent change in tumor burden per RECIST1.1 criteria (Supplemental Figure S1). We found no significant differences in the total aggregate PSI scores (representing all assayed cytokines) from CD4+ and CD8+ T cells between responders and non-responders (Supplemental Figures S2–S6).
Figure 1. CD8+ T-cell TNFα cytokine functionality and CD8+ T-cell p-STAT3 activity are associated with superior clinical response to transgenic TCR T-cell therapy.
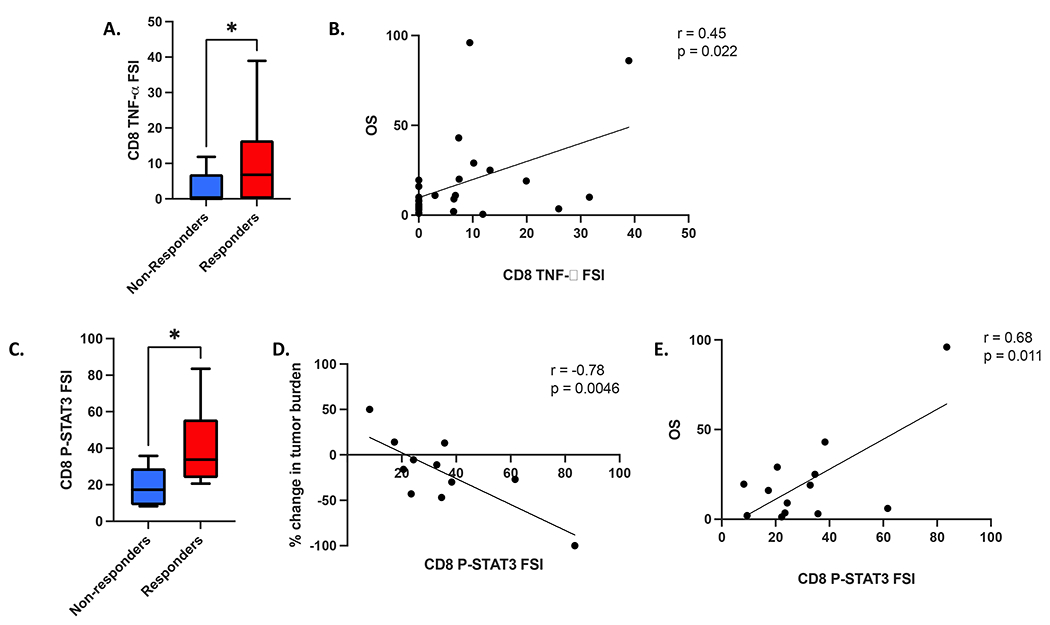
(A) CD8+ T-cell TNFα functional strength index (FSI), i.e. the percentage of single-cells secreting TNFα multiplied by the degree of cytokine secretion intensity, is significantly greater in clinical responders to therapy (n = 17) vs non-responders (n = 10). (B) CD8+ T-cell TNFα FSI is significantly associated with patient overall survival. (C) CD8+ T-cell tonic p-STAT3 FSI is significantly greater in clinical responders to therapy vs non-responders, and is significantly correlated with superior degree of antitumor response, as measured by peak percent change in tumor burden per RECIST1.1 criteria (D), as well as with greater patient overall survival (E). All box and whisker plots represent median at dividing line, with interquartile range represented by boxes, and lines representing minimum and maximum values for each dataset, and are compared by unpaired t-test (*p<0.05), while all correlation plots represent Pearson’s r values. Each sample was run as an independent experiment, and data were aggregated by clinical response for analysis.
In parallel, we conducted single-cell intracellular proteomics assays on an IsoPlexis/PhenomEx single-cell proteomics chip, which assays for both intracellular phospho-protein signaling pathway activity and intracellular cytokine levels simultaneously. Importantly, the intracellular cytokine levels are distinct from the secreted cytokine levels measured in the secretomics assays and are not directly comparable due to differences in the associated experimental assays as described above. Aggregate data for these experiments, including both tonic and stimulated conditions for responders and non-responders, are summarized in Supplemental Figures S7 and S8. Tonic phospho-STAT3 FSI in CD8+ T cells was significantly greater in clinical responders vs non-responders following NY-ESO-1 TCR-T cell therapy, and was also found to be positively correlated with both patient survival and peak change in tumor burden (Figure 1C–E). Tonic CD8+ T-cell phospho-NF-κB p65 FSI was also significantly associated with survival, while its association with peak change in tumor burden nearly achieved statistical significance (Supplemental Figure S9A–B). We also observed that tonic intracellular levels of IFNγ within the CD8+ T-cell compartment displayed significant correlations with peak change in tumor burden and patient survival (Supplemental Figure S9C–D), although conversely, we observed no significant correlations between IFNγ secreted cytokine functionality and clinical response, change in tumor burden, or patient survival (Supplemental Figure S10). Overall, the phosphoprotein FSI scores for p-IκBα, p-STAT3, and p-STAT5 correlated more strongly with intracellular cytokine FSI scores following antigen stimulation within the CD8+ T-cell compartment, compared to tonic conditions (Supplemental Figure S11, Supplemental Tables S1 and S2). The CD4+ T-cell compartment phosphoprotein FSI scores demonstrated more significant correlation between p-MEK1/2 activity and several intracellular cytokine FSI scores following antigen stimulation, compared to tonic conditions (Supplemental Figure S12, Supplemental Tables S3 and S4). However, none of these parameters showed any association with clinical response.
CD4+ Th2 cytokine activity is associated with inferior clinical response to TCR T cell therapy
When our observations on single-cell cytokine polyfunctionality and intracellular signaling activity were extended into the CD4+ T-cell compartment of the TCR-T cell therapy infusion products, we were able to classify different CD4+ Th cell groups by their component cytokine PSIs. Using this approach, we observed that the CD4+ Th2 cell PSI (i.e. the proportion of single cells secreting Th2 cytokines IL4, IL5, IL10, and IL13 multiplied by the signal intensity of those cytokines) was significantly greater in clinical non-responders to therapy vs responders (Figure 2A). Furthermore, we found that the CD4+ Th2 cell PSI was also positively associated with inferior peak changes in tumor burden/tumor growth (Figure 2B), and was also weakly correlated with lower peak %TCR+ cells in circulation following adoptive cell transfer (Supplemental Figure S13), but failed to achieve statistical significance (p = 0.08). When we conducted acellular cytokine analysis, subjecting the culture supernatants from the antigen stimulation co-cultures to the same multiplexed cytokine panels as we did at the single-cell level, we also observed that the Th2 cytokines IL-4 and IL-5 displayed a significant association with clinical non-response to therapy and were positively correlated with inferior peak change of tumor burden, i.e. tumor growth occurring following cell infusion (Supplemental Figure S14). Our observations at the single-cell phospho-proteome level showed that p-IκBα (a marker of NF-κB signaling activity) was significantly associated with the degree of CD4+ Th2 cell PSI, and was weakly associated with clinical non-response to NY-ESO-1 TCR-T cell therapy, although this did not achieve statistical significance (Supplemental Figure S15). We did not observe any significant associations between CD4+ Th2 cell PSI and patient survival (Supplemental Figure S16) or any associations between CD4+ Th1 cell cytokine polyfunctionality and clinical outcomes (Supplemental Figure S17). CD4+ Th9 cell functionality was weakly associated with clinical response to therapy but failed to achieve statistical significance (Figure 2A), while CD4+ Th17 cell functionality was significantly associated with patient survival (Supplemental Figure S18).
Figure 2. CD4+ Th2 cell cytokine polyfunctionality is associated with inferior clinical response to transgenic TCR T-cell therapy.
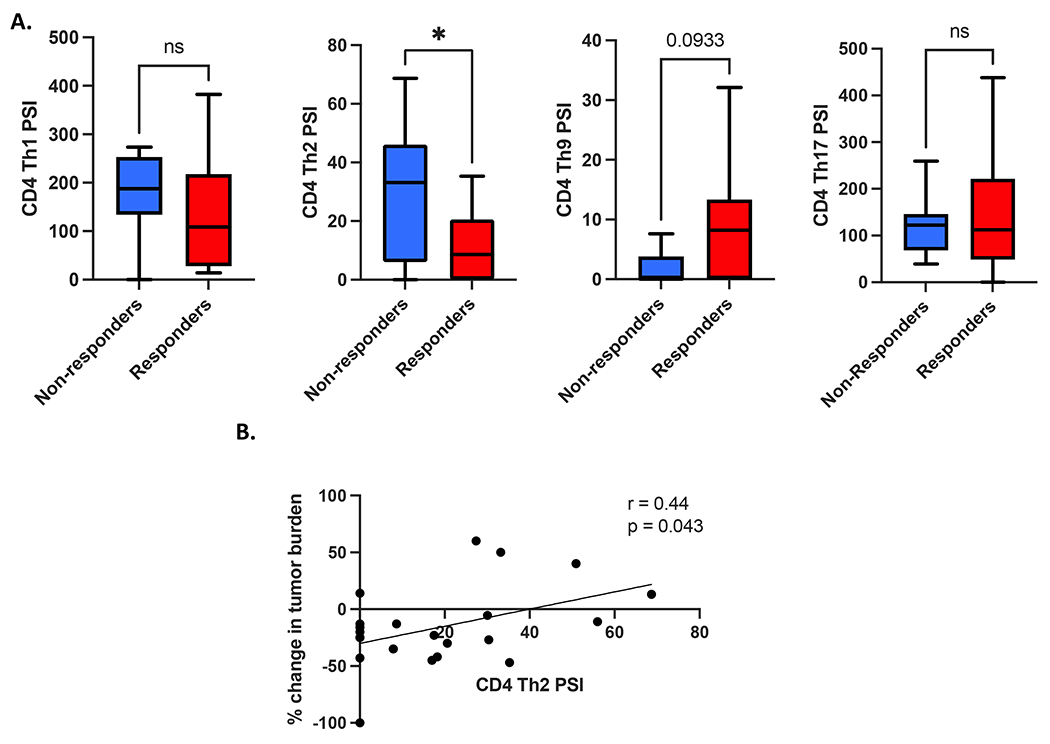
(A) Comparison of different CD4+ Th cell cytokine polyfunctionality profiles between clinical responders and non-responders to TCR T-cell therapy (Th1 = IL2, IFNγ, Th2 = IL4, IL5, IL10, and IL13, Th9 = IL9, Th17 = IL17A, IL17F, IL22). CD4+ Th2 cell polyfunctional strength index (PSI, i.e. proportion of single cells secreting Th2 cytokines IL4, IL5, IL10, and IL13 multiplied by the signal intensity of those cytokines) is significantly greater in clinical non-responders to therapy (n = 10) vs responders (n = 17). (B) CD4+ Th2 cell PSI is associated with inferior peak change in tumor burden per RECIST1.1 criteria. All box and whisker plots represent median at dividing line, with interquartile range represented by boxes, and lines representing minimum and maximum values for each dataset, and are compared by unpaired t-test (*p<0.05), while all correlation plots represent Pearson’s r values. Each sample was run as an independent experiment, and data were aggregated by clinical response for analysis.
Preinfusion patient serum cytokine levels associated with superior clinical response to TCR T cell therapy
In order to assess the impact of preinfusion serum cytokines on clinical responses to TCR T cell therapy, we performed 38-plex cytokine measurements on patient sera obtained on the day of cell product infusion (i.e., following conditioning chemotherapy), but prior to the cell infusion itself. We found that preinfusion serum levels of IL-15, Flt-3L, and CX3CL1 (fractalkine) were all significantly greater in responders compared to non-responders (Figure 3). Flt-3L was also weakly associated with superior degree of change in tumor burden but failed to fully achieve statistical significance (Supplemental Figure S19). We found no other significant associations with change in tumor burden, progression free survival, nor overall survival between these cytokines, nor any other cytokines assayed.
Figure 3. Preinfusion IL15, Flt-3L, and CX3CL1 serum levels are all associated with superior clinical response to transgenic TCR T-cell therapy.
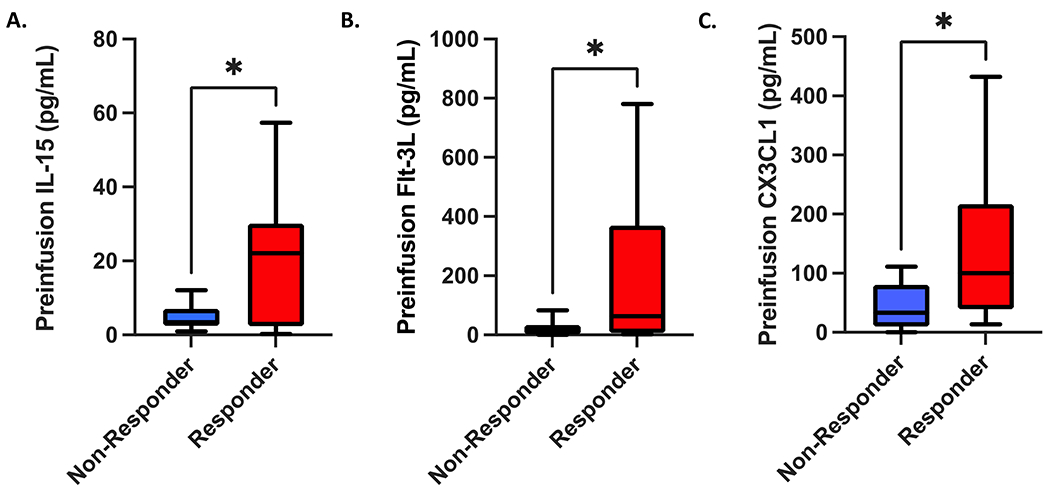
Serum levels of IL15 (A), Flt-3L (B), and CX3CL1/fractalkine (C) obtained prior to transgenic TCR T-cell infusions were all significantly greater in clinical responders to therapy (n = 17) compared to non-responders (n = 10). All box and whisker plots represent median at dividing line, with interquartile range represented by boxes, and lines representing minimum and maximum values for each dataset, and are compared by unpaired t-test (*p<0.05), while all correlation plots represent Pearson’s r values. Each sample was run as an independent experiment, and data were aggregated by clinical response for analysis.
DISCUSSION
TCR-T adoptive cell therapy has repeatedly demonstrated robust antitumor activity against solid tumors, but response durability and substantial numbers of non-responders to therapy remain urgent challenges for improvement. The single-cell characterization of T-cell cytokine polyfunctionality and signaling activity employed by our study, which we believe to be the first of its kind in clinical TCR T cell therapy products, enables greater resolution of specific cell populations and their cytokine producing/signaling activities that might be lost in traditional bulk assays. By interrogating the cytokine functionality and signaling elements associated with clinical response, and degree of clinical responsiveness to therapy, our exploratory, discovery-based studies has identified numerous potential targets for enrichment or exclusion in future generations of TCR T cell therapies for solid malignancies.
TNF-α is a pleiotropic cytokine with diverse roles in inflammation, immunity, and cell death. TNF-α enhances T-cell effector function, promotes T-cell survival, and improves antitumor immunity (10,11). TNF-α can also increase the sensitivity of tumor cells to apoptosis and promote the recruitment of immune cells into the tumor microenvironment in vivo (12–14). Therefore, the targeted enrichment of TNF-α secretion potential via CRISPR or TNF-α-“armed” vectors as an augmentation for TCR T-cell therapy could enhance T-cell function and improve antitumor immunity in solid tumors (15). Furthermore, STAT3 signaling activities have been shown to be associated with improved CD8+ T-cell effector functionality and antitumor efficacy in vivo, as well as playing a critical role in the development and maintenance of CD8+ memory T cells (16,17), which are vitally important for responsiveness to cell therapeutics. The importance of NF-κB signaling in this latter process has also been well described (18). Given the previously described deleterious impact of prior cumulative chemotherapy exposure on T-cell phenotypes and their potential for use in adoptive cell therapies (19), it may be that more refined screening for certain cell signaling pathway activities, and their subsequent enrichment, may be of benefit to future generations of adoptive cell therapies.
CD4+ Th2 cell immune responses are characterized by the production of the cytokines IL-4, IL-5, IL-10, and IL-13. While important for the defense against extracellular pathogens and allergic responses, Th2 cytokines are also known to be immunosuppressive in the setting of T cell–directed immunity, and can inhibit the anticancer functionality of T cell–based immunotherapies (20–22). While a previous study demonstrated a positive association between CD4+ Th2 cell levels and lack of clinical relapse in anti-CD19 CAR T-cell therapy for acute lymphoblastic leukemia (23), our study utilized samples from a greater number of patients and focused on solid malignancies instead of leukemia. Other studies have demonstrated the importance of CD4+ Th9 and Th17 cells and their associated cytokines with clinical response to both TCR T-cell and CAR T-cell therapies in vivo (3,24). Although the importance of CD4+ Th1 cells in adoptive cell therapy approaches has been well described (25), we did not observe any association between Th1 polyfunctionality and clinical response. It is plausible that the impact of CD4+ Th cell cytokine functionality is highly dependent on both the type of tumor microenvironment as well as the type of transgenic T cell being utilized, as TCR T cells and CAR T cells display differing degrees of antigen-dependent activity in vivo. Selective depletion of CD4+ Th2 cell components of TCR T cell products may therefore be an effective way to augment their effectiveness in vivo.
The importance of circulating cytokines to ACT performance in vivo, as well the degree to which conditioning chemotherapy is able to augment them, remains an important and actionable area of study. The positive impact of higher levels of serum IL-15 (as a consequence of conditioning chemotherapy efficacy) on CAR T-cell clinical outcomes has been well described (3,4). CX3CL1 (fractalkine) signaling improves the antitumor efficacy of T-cells in vivo (26), but its role in transgenic adoptive cell therapy has been less well characterized. Transgenic Flt-3L has been utilized to augment epitope spreading and antitumor activity of adoptively transferred T cells in vivo (27), but has not been explored as a surrogate for enhancing clinical adoptive cell therapy performance. It is possible that these and other factors can be utilized as either clinical biomarkers to help predict adoptive cell therapy outcomes, stratify effectiveness of different conditioning chemotherapy regimens, or as targets for supplementation and enrichment in patients who are to receive these therapies.
Our exploratory study has several limitations. One major limitation is the inability to fully assess the changes experienced in cytokine polyfunctionality and signaling activity over time in the patients, as obtaining sufficiently large quantities of tumor-infiltrating lymphocytes from on-treatment tumor biopsies was not feasible. However, previous studies have shown the importance of post-infusion IFN-γ and IL-6 levels for clinical response to TCR T-cell therapeutics (28). Furthermore, obtaining on-treatment tumor biopsies in order to interrogate the patients’ tumor microenvironment were not feasible to obtain, as many patients’ metastases were not surgically accessible. This limited our ability to adequately assay their impact on systemic responses, as well as assessing the impact of the various Th cell subtypes within the tumor microenvironment. Finally, our culture conditions utilized IL-2, which is generally necessary for the successful ex vivo culture of primary human T cells. This may have introduced HLA-independent, NK cell–like activity in the T cells. Indeed, we observed high granzyme B secretion under tonic conditions, as well as the secretion levels of several other cytotoxic cytokines (MIP-1α, MIP-1β). This may indeed be due to increased HLA-independent T-cell responses, as IL-2 has been shown to induce these responses independent of antigen stimulation (29). It is also known that IL-2 is able to impact signaling through STAT1, STAT3, and STAT5 (30), so IL-2 culture conditions may have impacted the tonic level of signaling through these pathways. However, given that our manufacturing protocols utilized anti-CD3 and IL-2 mediated expansion, and that all patients received systemic IL-2 for 1-2 weeks following adoptive cell transfer (1,2), it is likely that any IL-2-mediated T-cell activity that we observed would still be reflective of what occurs in vivo.
It is likely that chronic antigen stimulation, as well as sustained exposure to different degrees of local cytokine stimulation within the tumor microenvironment, can impact the cytokine functionality and intracellular signaling pathway activity of the adoptively transferred TCR T cells. For example, in addition to its impact on T-cell effector functionality and direct antitumor activity, TNF-α signaling via TNF-receptor 2 (TNFR2) and associated canonical NF-κB signaling activity has been shown to directly inhibit Th2 cytokine production, can also inhibit the differentiation of Th2 cells in vitro (31,32), which is consistent with our data showing that NF-κB activity (as measured by p-IκBα activity) was positively associated with CD4+ Th2 cell cytokine polyfunctionality. It is possible that TNF-α activity, as well as signaling by other inflammatory cytokines such as interferon-gamma through JAK/STAT pathways, may augment or even potentiate divergences in local T-cell phenotype and functionality in the tumor microenvironment in vivo. Further mechanistic studies both in vitro and in vivo, as well as larger, prospective clinical cohorts with access to on-treatment biopsy samples are necessary to comprehensively address the complexities of all of the factors studied herein in the future.
In summary, we have shown that superior clinical responses to transgenic TCR T-cell therapies are characterized by increased CD8+ T-cell TNFα cytokine functionality and phospho-STAT3 activity, and by decreased CD4+ Th2 cell cytokine activity. Furthermore, we have demonstrated that increased preinfusion serum IL15, Flt-3L, and CX3CL1 levels are also associated with superior clinical responsiveness to therapy. These findings have implications in how the cellular therapeutics community should approach the design of future generations of these products, both in terms of vector design, cell product enrichment strategies, and conditioning chemotherapy for patients treated with these agents for solid tumors.
Supplementary Material
SYNOPSIS:
The authors show that differences in the cytokine profiles of transgenic cellular immunotherapies for solid tumors have implications for therapeutic efficacy. These differences should be considered when developing future generations of cellular therapies to augment clinical response rates.
ACKNOWLEDGEMENTS:
We gratefully acknowledge Tiffany Coupet, Edward Han, and Olivia Loisel of PhenomeX (formerly IsoPlexis) for technical assistance with the single-cell intracellular phospho-proteome experiments and data analysis.
GRANT SUPPORT:
T.S.N. is supported by the NIH grants K08 CA 241088 and P30 CA016042, the California Institute for Regenerative Medicine (CIRM) grant CLIN2-11380, the Hyundai Hope on Wheels Hope Scholar Award, funding from the UCLA Jonsson Comprehensive Cancer Center, and the Ruby Family Foundation. A.R. is supported by the Parker Institute for Cancer Immunotherapy (PICI), the NIH grants R35 CA197633 and P01 CA244118, the Ressler Family Fund, Ken and Donna Schultz, Todd and Donna Jones, and Thomas Stutz.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST: T.S.N. has received honoraria from consulting with Allogene Therapeutics. A.R. has received honoraria from consulting with Amgen, Bristol-Myers Squibb, Chugai, Genentech, Merck, Novartis, Roche and Sanofi, is or has been a member of the scientific advisory board and holds stock in Advaxis, Arcus Biosciences, Bioncotech Therapeutics, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Lutris Pharma, Merus, PACT Pharma, Rgenix and Tango Therapeutics, and has received research funding from Agilent and Bristol-Myers Squibb through Stand Up to Cancer (SU2C). B.C-A. has received honoraria from consulting with Advarra IBC. The rest of the authors declare no potential conflicts of interest.
DISCLAIMER: The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the NIH, the NCI, Hyundai Hope on Wheels, the Parker Institute for Cancer Immunotherapy, the Ruby Family Foundation, or the Ressler Family Fund.
REFERENCES
- 1.Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res 2014;20(9):2457–65 doi 10.1158/1078-0432.CCR-13-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, et al. A Pilot Trial of the Combination of Transgenic NY-ESO-1-reactive Adoptive Cellular Therapy with Dendritic Cell Vaccination with or without Ipilimumab. Clin Cancer Res 2019;25(7):2096–108 doi 10.1158/1078-0432.CCR-18-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells associate with clinical outcomes in NHL. Blood 2018. doi 10.1182/blood-2018-01-828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 2008;26(32):5233–9 doi 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma C, Cheung AF, Chodon T, Koya RC, Wu Z, Ng C, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discov 2013;3(4):418–29 doi 10.1158/2159-8290.CD-12-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45(2):228–47 doi 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 7.Britten CM, Meyer RG, Kreer T, Drexler I, Wolfel T, Herr W. The use of HLA-A*0201-transfected K562 as standard antigen-presenting cells for CD8(+) T lymphocytes in IFN-gamma ELISPOT assays. J Immunol Methods 2002;259(1-2):95–110 doi 10.1016/s0022-1759(01)00499-9. [DOI] [PubMed] [Google Scholar]
- 8.Tang JP, Peters CW, Quiros C, Wang X, Klomhaus AM, Yamada RE, et al. Hypophosphatemia Due to Increased Effector Cell Metabolic Activity Is Associated with Neurotoxicity Symptoms in CD19-Targeted CAR T-cell Therapy. Cancer Immunol Res 2022;10(12):1433–40 doi 10.1158/2326-6066.CIR-22-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comin-Anduix B, Gualberto A, Glaspy JA, Seja E, Ontiveros M, Reardon DL, et al. Definition of an immunologic response using the major histocompatibility complex tetramer and enzyme-linked immunospot assays. Clin Cancer Res 2006;12(1):107–16 doi 10.1158/1078-0432.CCR-05-0136. [DOI] [PubMed] [Google Scholar]
- 10.Laha D, Grant R, Mishra P, Nilubol N. The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front Immunol 2021;12:656908 doi 10.3389/fimmu.2021.656908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyciakova S, Valova V, Svitkova B, Matuskova M. Overexpression of TNFalpha induces senescence, autophagy and mitochondrial dysfunctions in melanoma cells. BMC cancer 2021;21(1):507 doi 10.1186/s12885-021-08237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Loo G, Bertrand MJM. Death by TNF: a road to inflammation. Nat Rev Immunol 2023;23(5):289–303 doi 10.1038/s41577-022-00792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, et al. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J Clin Invest 2007;117(12):3833–45 doi 10.1172/JCI32567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine 2018;101:14–8 doi 10.1016/j.cyto.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freeman AJ, Kearney CJ, Silke J, Oliaro J. Unleashing TNF cytotoxicity to enhance cancer immunotherapy. Trends Immunol 2021;42(12):1128–42 doi 10.1016/j.it.2021.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Sun Q, Zhao X, Li R, Liu D, Pan B, Xie B, et al. STAT3 regulates CD8+ T cell differentiation and functions in cancer and acute infection. J Exp Med 2023;220(4) doi 10.1084/jem.20220686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deenick EK, Pelham SJ, Kane A, Ma CS. Signal Transducer and Activator of Transcription 3 Control of Human T and B Cell Responses. Front Immunol 2018;9:168 doi 10.3389/fimmu.2018.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes SE, Wang Y, Chen L, Molinero LL, Gajewski TF, Evaristo C, et al. T cell-NF-kappaB activation is required for tumor control in vivo. J Immunother Cancer 2015;3(1):1 doi 10.1186/s40425-014-0045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov 2019;9(4):492–9 doi 10.1158/2159-8290.CD-18-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454(7203):436–44 doi 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 21.Protti MP, De Monte L. Cross-talk within the tumor microenvironment mediates Th2-type inflammation in pancreatic cancer. Oncoimmunology 2012;1(1):89–91 doi 10.4161/onci.1.1.17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell reports 2017;20(3):757–70 doi 10.1016/j.celrep.2017.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai Z, Woodhouse S, Zhao Z, Arya R, Govek K, Kim D, et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci Adv 2022;8(23):eabj2820 doi 10.1126/sciadv.abj2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Y, Chen J, Bi E, Zhao Y, Qin T, Wang Y, et al. TNF-alpha enhances Th9 cell differentiation and antitumor immunity via TNFR2-dependent pathways. J Immunother Cancer 2019;7(1):28 doi 10.1186/s40425-018-0494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li K, Donaldson B, Young V, Ward V, Jackson C, Baird M, et al. Adoptive cell therapy with CD4(+) T helper 1 cells and CD8(+) cytotoxic T cells enhances complete rejection of an established tumour, leading to generation of endogenous memory responses to non-targeted tumour epitopes. Clin Transl Immunology 2017;6(10):e160 doi 10.1038/cti.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siddiqui I, Erreni M, van Brakel M, Debets R, Allavena P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J Immunother Cancer 2016;4:21 doi 10.1186/s40425-016-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol 2020;21(8):914–26 doi 10.1038/s41590-020-0676-7. [DOI] [PubMed] [Google Scholar]
- 28.Gyurdieva A, Zajic S, Chang YF, Houseman EA, Zhong S, Kim J, et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nature communications 2022;13(1):5296 doi 10.1038/s41467-022-32491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamang DL, Redelman D, Alves BN, Vollger L, Bethley C, Hudig D. Induction of granzyme B and T cell cytotoxic capacity by IL-2 or IL-15 without antigens: multiclonal responses that are extremely lytic if triggered and short-lived after cytokine withdrawal. Cytokine 2006;36(3-4):148–59 doi 10.1016/j.cyto.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross SH, Cantrell DA. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol 2018;36:411–33 doi 10.1146/annurev-immunol-042617-053352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng J, Li XM, Zhang GR, Cheng Y, Chen X, Gu W, et al. TNF-TNFR2 Signaling Inhibits Th2 and Th17 Polarization and Alleviates Allergic Airway Inflammation. Int Arch Allergy Immunol 2019;178(3):281–90 doi 10.1159/000493583. [DOI] [PubMed] [Google Scholar]
- 32.Li XM, Chen X, Gu W, Guo YJ, Cheng Y, Peng J, et al. Impaired TNF/TNFR2 signaling enhances Th2 and Th17 polarization and aggravates allergic airway inflammation. Am J Physiol Lung Cell Mol Physiol 2017;313(3):L592–L601 doi 10.1152/ajplung.00409.2016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its Supplementary Data Files or upon request from the corresponding author.