

Abstract
Purpose:
Speckle-type POZ protein (SPOP) is important in DNA damage response (DDR) and maintenance of genomic stability. Somatic heterozygous missense mutations in the SPOP substrate-binding cleft are found in up to 15% of prostate cancers. While mutations in SPOP predict for benefit from androgen receptor signaling inhibition (ARSi) therapy, outcomes for patients with SPOP-mutant (SPOPmut) prostate cancer are heterogeneous and targeted treatments for SPOPmut castrate-resistant prostate cancer (CRPC) are lacking.
Experimental Design:
Using in silico genomic and transcriptomic tumor data, proteomics analysis and genetically modified cell line models we demonstrate mechanistic links between SPOP mutations, STING signaling alterations and PARP inhibitor vulnerabilities.
Results:
We demonstrate that SPOP mutations are associated with upregulation of a 29-gene non-canonical (NC) STING (NC-STING) signature in a subset of SPOPmut, treatment-refractory CRPC patients. We show in preclinical CRPC models that SPOP targets and destabilizes STING1 protein, and prostate cancer – associated SPOP mutations result in upregulated NC-STING-NF-κB signaling and macrophage- and tumor microenvironment (TME)-facilitated reprogramming, leading to tumor cell growth. Importantly, we provide in vitro and in vivo mechanism-based evidence that PARP inhibitor (PARPi) treatment results in a shift from immunosuppressive NC-STING-NF-κB signaling to anti-tumor, canonical cGAS-STING-IFN-β signaling in SPOPmut CRPC and results in enhanced tumor growth inhibition.
Conclusions:
We provide evidence that SPOP is critical in regulating immunosuppressive versus anti-tumor activity downstream of DNA damage-induced STING1 activation in prostate cancer. PARPi treatment of SPOPmut CRPC alters this NC-STING signaling toward canonical, anti-tumor cGAS-STING-IFN-β signaling, highlighting a novel biomarker-informed treatment strategy for prostate cancer.
INTRODUCTION
Mutations in DNA damage response (DDR) genes are seen in upwards of 30% of metastatic prostate cancers, with multiple PARP inhibitors (PARPis) having approved indications in men with metastatic castrate-resistant prostate cancer (CRPC) with mutations in genes involved in homologous recombination repair (HRR) (1). Speckle-type POZ protein (SPOP) is a substrate adaptor of cullin3 (Cul3)-RING ubiquitin ligase, and somatic heterozygous missense mutations in the SPOP substrate binding cleft are found in up to 15% of prostate cancers (2). Prominent, recurrent prostate cancer–associated missense SPOP mutations were shown to reduce SPOP substrate ubiquitination include Y87C/N, F102C/V, F125L, W131G, and F133V/L, with F133V/L being the most prevalent SPOP mutation identified. These mutations occur in the N-terminal MATH domain of SPOP, which recognizes SPOP-binding consensus (SBC) motifs on the substrate (3,4). SPOP was shown to regulate genomic stability through modulation of DNA double-strand break (DSB) repair in prostate cancer (5) and by promoting expression of DNA repair and replication factors to mitigate genomic instability (6). SPOP-mutant (SPOPmut) prostate cancer cells have been shown to be sensitive to PARPis in vitro, yet clinical studies dedicated to assessing the predictive value of SPOP mutations in precision therapy approaches are lacking (5,6) with no formal prospective analysis of PARPi in patients with SPOP mutant mCRPC. Clinical benefit to PARPi has been seen in prostate cancers with mutations in other DDR genes beyond just BRCA1/2 (7,8). There are many published genomic signatures of homologous recombination repair deficiency that rely on identifying genomic features of BRCAness (9). A recent study of one HRD signature in a small number of prostate tumors did not show the presence of the HRD signature in SPOP-mutant tumors; however, it is important to note that these genomic signatures thus far have failed to identify HRR-proficient tumors that benefit from PARPi, and certain gene mutations, such as in ATM, do not display these BRCAness signatures but response to PARPi-based treatments can be seen (9). Although SPOP mutations predict for prolonged benefit to ARSi in patients (10), strategies to more specifically target SPOPmut castration-resistant prostate cancer (CRPC) are needed.
Recent studies have identified canonical STING1 signaling, integrated as the anti-tumor canonical cGAS-STING-TBK1-IRF3 innate immune response pathway in many cancer types including prostate cancer, as an essential mechanism that enhances the sensitivity of cancers to DDR inhibitor (DDRi)-induced growth inhibition and cell death (11,12). Mechanistically, this depends on the accumulation of cytosolic DNA from the chromosomal DNA that is unrepaired due to DNA damaging treatments. The accumulated cytosolic DNA activates cGAS for production of cGAMP to activate STING1. Our previous publication showed that DDRis (PARPi and ATRi) induce DNA damage and cytoplasmic double-strand DNA (dsDNA)-mediated canonical STING signaling (cGAS-STING-IFN-β), leading to downstream intrinsic IFN-β–mediated cytotoxicity, reprogramming of the tumor microenvironment (TME), and T cell–dependent and –independent anti-tumor activity in DDR–wild-type CRPC models (13). Interestingly, without the cytosolic activities of cGAS/cGAMP, etoposide-induced DNA damage and intranuclear DDR signaling (involving IFI16 and DDR factors ATM and PARP1) can directly activate STING1, resulting in activation of pro-tumor growth, non-canonical STING (NC-STING) with downstream induction of an NF-κB transcriptional program (14). Constitutive activation of NF-κB was observed in many types of cancers by transcriptionally targeting the expression of multiple genes, such as certain cytokines (e.g., IL-1, IL-2, IL-6, IL-8, IL-15) (15–18) and antiapoptotic (prosurvival) BCL-2 family members and caspase inhibitors (e.g., BCL2 and BCL-XL) (19,20). Specifically in prostate cancers, NF-κB activation is reportedly involved in carcinogenesis and castration-resistant progression by inducing the expression of multiple protein factors and pathways, including IL-6, IL-23, and other signaling pathways involving angiogenesis, invasion, and metastasis (21,22).
Thus, STING1 signaling–induced NF-κB may mechanistically drive the development, survival, and proliferation of CRPC, yet at a fundamental level the alternative regulation of this pro- versus anti-tumor signaling downstream of STING1 activation is unclear. In particular, the inter-regulatory interactions between NC-STING-NF-κB signaling and canonical STING1 signaling within the context of DDRi treatment is unknown, yet a mechanistic understanding of these interactions would be of paramount importance in the stratification of patients with prostate cancer for targeted therapies. Herein, we identified STING1 protein as an SPOP-binding substrate for degradation through SPOP-mediated ubiquitin-proteasome pathway, and demonstrate that prostate cancer – associated SPOP mutations stabilize STING1 to amplify NC-STING-NF-κB signaling, but also create a therapeutic vulnerability to PARP inhibition whereby the NC-STING1 signaling is altered to promote canonical STING-IFN-β–mediated tumor cell killing and increased apoptosis.
METHODS
Expression analysis
For RNA-seq data analysis, the raw RNA-seq sequence reads were aligned using STAR RNA-Seq aligner (STAR, Version 2.7.10b). mRNA transcripts in each sample were quantified, normalized and further analyzed for differential expression analysis as previously described (13), and detailed in Supplementary Methods. Functional evaluation of the transcriptomic data for TNF-α-NF-κB signaling pathway or a compound STING-NF-κB signaling pathway 259-gene set was performed using a customized gene set collection defined in relevant existing literature (Supplementary Table S1). Pre-ranked GSEA was performed based on a test statistic obtained from differential expression between SPOPmut and SPOPwt cohorts. NC_STING_score was computed as z-score of gene expression of NC-STING signature derived from the Beltran cohort.
Proteomic profiling of SPOPwt and SPOPmut prostate cancer models
Dox-inducible SPOPwt (EV) and SPOPmut (F102C and F133V) C4–2b prostate cancer models were labeled with 13C6 Lys (#CNLM-2247, Cambridge Isotope Laboratories) in RPMI1640 containing 10% dialyzed FBS and 1% penicillin/streptomycin cocktail (Gibco). The cells were collected, lysed, sonicated and fractioned for LC-MS/MS analysis as detailed description in Supplementary Methods. The generated MS/MS spectra were searched against the Uniprot database (Human and Bovine, January 2017) using the X!Tandem search engine through the Trans-Proteomic Pipeline (TPP 4.8) and processed with the Peptide and Protein Prophet. Trypsin was specified as protein cleavage site, with the possibility of two missed cleavages allowed. For the modifications, one fixed modification of propionamide (71.037114) at cysteine and two variable modifications, oxidation at methionine (15.9949 Da) and SILAC 13C6 at lysine (6.0201 Da), were chosen. Addition of SILAC was used strictly to discriminate human protein from bovine protein and was not intended to perform relative quantitation of Heavy versus Light ratios. The mass error allowed was 10 ppm for parent monoisotopic and 0.5 Da for MS2 fragment monoisotopic ions. The searched result was filtered with FDR = 0.01. Ingenuity Pathway Analyses of these 81 proteins revealed enriched representation of an NF-κB–centric protein network (Supplementary Table S2).
Cell lines and cell culture reagents
Parental RM-1-BM mouse prostate cancer cell line—derived from a ras+myc–induced mouse prostate cancer tumor described previously (13)—were obtained from Dr. Carl Powers, NSW Australia and maintained in DMEM cell culture medium (Cat# D6046, Sigma-Aldrich, MO) supplemented with 10% fetal bovine serum (FBS). Parental LNCaP C4–2b cells (obtained from Dr. Gary E. Gallick, MD Anderson) were maintained in RPMI 1640 cell culture medium (Cat# R7509, Sigma-Aldrich) with 10% FBS.
RM-1-BM cells lines with stably transfected Dox-inducible lentiviral vectors pInducer-EV pInducer-HA-tagged SPOPwt, pInducer-SPOPF102C or pInducer-SPOPF133V were maintained in DMEM culture medium with 10% FBS and 200 ug/mL G-418 (Cat# G-1033, AG Scientific, CA). Dox-inducible C4–2b cell lines (pInducer-EV, pInducer-HA-tagged SPOPwt, pInducer-HA-tagged SPOPF102C, or pInducer-HA-tagged SPOPF133V) were maintained in RPMI 1640 cell culture medium supplemented with 10% FBS and 200 μg/mL G-418. Human embryo kidney 293T cells were obtained from ATCC (Cat# CRL-3216) and maintained in DMEM culture medium containing 10% FBS. RM-1-BM or C4–2b cell lines with stably infected pLenti-EV or pLenti-HA-tagged SPOPs (HA-SPOPwt, HA-SPOPF102C, or HA-SPOPF133V) were cultured and maintained in DMEM or RMPI1640 supplemented with 10% FBS plus 0.5 μg/mL puromycin (Cat# P9620, Sigma-Aldrich), respectively. Cell lines are aliquoted at early passage number, aliquoted and stored in liquid nitrogen freezers until use. Cells are passaged 4–5 times during experiments and early passage cells are revived if needed. Cell lines are authenticated by short tandem repeat DNA fingerprinting with the AmpFlSTR Identifier PCR Amplification Kit (ThemoFisher) and each derived cell line was tested for mycoplasma using Lonza Mycoalert Mycoplasma Detection Kit, at MD Anderson Cytogenetics and Cell Authentication Core.
PARPis [olaparib (AZD2281) and talazoparib (BMN 673), Cat# S1060 and S7048, respectively] and 20S proteasome inhibitor PS-341 (bortezomib, Cat# S1013) were obtained from Selleck Chemicals (TX). Protein synthesis inhibitor cycloheximide (for CHX chase assays) was from Sigma-Aldrich (Cat# C7698). Virus infection reagent polybrene (hexadimethrine bromide, Cat# H9268, Sigma-Aldrich) was dissolved in nuclease-free water (16 mg/mL), sterilized by 0.22-μm filtration, and stored at −80°C. Anti-human (Cat# mabg-hil6–3) or anti-mouse (Cat# mabg-mil6–3) interleukin 6 (IL-6) neutralization antibodies, Rat IgG2a Control (Cat# mabg2a-ctlrt, control IgGs for anti-mIL-6 Abs) and Mouse Control IgG1 (Cat# mabg1-ctlrt, control IgGs for anti-hIL-6 Abs) were from Invivogen (CA). Anti-mouse (Cat# 32400) and anti-human (Cat# 31410) IFN-β neutralization antibodies were purchased from PBL Assay Science, NJ. Matrigel® (Cat# 354248) purchased from Corning Life Sciences, NY.
Coculture models
Mouse (RAW264.7) and human (THP-1) macrophage cells were obtained from ATCC (Cat#s TIB-71 and TIB-202, respectively) grown in 24-well cell culture plates and cocultured with RM-1-BM (VC, SPOPF102C, and SPOPF133V) or C4–2b (VC, SPOPF102C, and SPOPF133V) prostate cancer cell line models plated in 0.4-μm TransWell cell culture inserts (Cat# 08–770, ThermoFisher), respectively. The plates were incubated in 37°C CO2 incubator for 24 hrs before treatment with the indicated drugs for 48 hrs. The treated cells (macrophage and prostate cancer cells with or without coculture)] were analyzed by MTS assay. For immunoblot (IB) and RT-qPCR analysis, after the treatment, the cells were collected and extracted for whole cell proteins or total RNA. The detailed methods and materials are provided in Supplementary Methods.
Plasmid constructs, lentivirus packaging, infection and establishment of stably transfected SPOP-expressing prostate cancer model cell lines
Venus1-tagged STING1 (STING1-V1) was a gift from Eric Schirmer (Addgene plasmid #124262).
Plasmid constructs that were used in in vivo ubiquitination assay, in vitro overexpression analysis and to establish stably transduced prostate cancer model cell lines are described in Supplementary Methods, together with the source of materials, cloning methods including mutagenesis procedures and primers.
To prepare lentivirus-containing SPOP expression vectors or EV, pInducer-SPOPs, pInducer-EV, pLenti-SPOPs, or pLenti-EV were co-transfected with packaging plasmid constructs into 293T cells utilizing Lipofectamine 2000 (Cat# 11668019, ThermoFisher). Forty-eight hours after transfection, the lentiviral supernatant (containing respective pInducer-EV/SPOPs or pLenti-EV/SPOPs virus) was collected, sterile-filtered (0.45 μm) and used for cell infection as described previously (23).
To infect RM-1-BM or C4–2b parental cells, filtered viruses were added to the culture incubation containing 8 μg/mL polybrene to infect the cells. Forty-eight hours after infection, the cultures were changed to fresh media and incubated for another 24 hours to expand the infected cell cultures before G418 (pInducer-EV/SPOP infected lines, 700 μg/mL for C4–2b and 1000 μg/mL for RM-1-BM) or puromycin (pLenti-EV/SPOP infected lines, 4 μg/mL for C4–2b or RM-1-BM) was introduced to select the stably infected clones. The resultant stably infected prostate cancer model cell lines were confirmed, characterized [inducible capability (pInducer-SPOPs) or constitutive expression of exogenous SPOPs (pLenti-SPOPs) carried by lentiviral vectors], and used in our designated experiments.
Immunoblotting analysis
Cells were washed with phosphate-buffered saline (PBS, pH7.4) and whole-cell proteins were extracted by 1x RIPA lysis buffer (Cat# 9806, Cell Signaling Tech, MA) plus proteinase inhibitor cocktail (Cat# 4693132001, Roche, MilliporeSigma, MA) and phosphatase inhibitor cocktail (Cat# 4906837001 PHOSS-RO, MilliporeSigma). The cell lysates were sonicated and centrifuged to remove debris. IB analysis was performed following standard IB protocols described in our previous publications (13,23,24). Briefly, equal amounts of total proteins were loaded and separated by precast gels (Cat# 4561086, Bio-Rad, CA; or Cat# M00657, GenScript, NJ) and transferred to Immun-Blot® PVDF membranes (Cat# 1620177, Bio-Rad). The membranes were blocked by nonfat milk and incubated with primary antibodies, followed by incubation with HRP-conjugated secondary antibodies (goat anti-rabbit HRP, Cat# 31460 and goat anti-mouse HRP, Cat# 31430, Thermo Fisher Scientific). The protein blotting signals were visualized by incubating the membranes with SuperSignal™ West substrates (Cat# 34580, Cat# 37071, or Cat# 34096, Thermo Fisher Scientific) and scanned by a VersaDoc™ MP Image System (Bio-Rad). The protein blotting images were obtained, processed, and quantified by Image Lab™ (Ver. 6.0.1 build 34, Bio-Rad). All antibodies used in IB analysis are listed in Supplementary Table S3.
Immunostaining analysis and quantitation
For immune- and immunofluorescence stain analysis, paraffin tissue sections were de-paraffinized, dehydrated and subjected to antigen retrieval as previously described and detailed in Supplementary Methods.
For immunostaining, slides were stained with protein specific antibodies (Supplementary Table S3) as described in Supplementary Methods, and mounted with Permount Mounting Media (Cat# SP15–500, ThermoFisher) and were then observed and images taken with a light microscope (Nikon Eclipse 90i).The antibody specific DAB staining intensity was quantitated by using ImageJ from 5 random hot areas in one representative well white-balanced image of, at least, three different fields of one tumor sample. Two to three tumors from each group were analyzed. The DAB specific staining intensity was calculated with the following formula: DAB specific staining intensity=Log (max intensity/mean intensity) where max intensity = 255 for 8-bit images.
For immunofluorescence and double immunofluorescence analysis, slides were incubated with protein specific antibodies followed by fluorescent dye-labeled secondary antibodies as described in Supplementary Methods. The slides were mounted with Prolong antifade with DAPI (Cat#P36931, ThermoFisher) and immunofluorescence was visualized and images captured utilizing a Leica SP8 fluorescent microscope. Slides from representative tumor from each group was used and one representative image from, at least, five different fields with tumor cells from one slide was presented. Total number of cells were assessed with ImageJ. For double immunofluorescence analysis, numbers of cells with clear cleaved caspase-3, PARP1 cytoplasmic accumulation, and both were counted manually. All primary antibodies used in immunostaining analysis are listed in Supplementary Table S3 and Supplementary Methods.
RT-qPCR analysis
Total RNA in cells was isolated by TRIzol Reagent (Cat# 15596026, ThermoFisher) and reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Cat# 4368813, ThermoFisher). RT-qPCR was conducted with Fast SYBR Green Master Mix (Cat# 4385610, ThermoFisher) on a StepOnePlus™ Real-Time PCR System (ThermoFisher). 2-ΔΔCt method was used to measure relative mRNA expression compared to different treatment conditions or controls. The qPCR primers to detect specific gene transcripts are designed and synthesized by custom DNA oligos synthesis services (ThermoFisher), and the primer sequences are listed in Supplementary Table S4.
Cycloheximide chase assay to determine protein half-life
Briefly, CHX was added to the cell incubation at the final concentration of 100 ug/mL. The cells were lysed, and cell lysates collected at each elapsed time point, designated at 0, 0.5, 1, 2.5, 5, and 10 hours, following the introduction of CHX incubation. The proteins in the cell lysates were separated by PAGE, transferred to membranes immunoblotted with protein-specific antibodies, visualized, and imaged as we described in “Immunoblotting analysis” above. The quantified protein blot signals were normalized to the blot signal of vinculin within each sample (as internal protein loading control). The normalized protein blot signal from each time point (0.5, 1, 2.5, 5, and 10 hours) was then calculated as folds of the normalized protein blot signal at 0 hours (initiation control, as 1 fold) and plotted to display the time-elapsed presentation of proteins (folds of the 0 hour time point) following CHX treatment to block the initiation of new protein synthesis. Half-life of the protein is read as the approximate hour when 0.5 fold of protein is observed compared to 0 hour in the plotted figure.
Plasmid DNA transfection in mammalian cells
293T cells were transfected with expression vectors using Lipofectamine 2000 transfection reagent (Cat# 11668019, ThermoFisher) following the manufacturer’s instructions. Typically, for a 6-well plate, 3 μg total plasmid DNA per well was transfected, and for a 10-cm plate, 10 μg of total plasmid DNA was optimized for the transfection. Forty-eight hours post transfection, the cells were harvested and washed with PBS. Total cell lysates or total RNAs were extracted and used for Co-IP, immunoblot, or RT-qPCR analysis as described above in “Immunoblotting analysis” and “RT-qPCR analysis” for the indicated experiments.
Co-Immunoprecipitation (Co-IP) analysis
For in vitro analysis of protein-protein interactions, protein expression vectors (HA-tagged SPOP, pcDNA3.1-HA-SPOP; Venus-tagged STING1, STING1-V1) or empty plasmid DNA vectors (total DNA transfection control) were mono- or co-transfected (as indicated) into 293T cells. Forty hours after the transfection, the cells were collected, washed by PBS, and lysed in NP-40 lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM DTT, 1 mM PMSF] containing protease inhibitor cocktail (Cat# 4693132001 Roche, MilliporeSigma) and phosphatase inhibitor cocktail (Cat# 4906837001 PHOSS-RO, MilliporeSigma). Cell lysates were cleared by centrifuge and quantified. The same amount of total proteins from each sample was incubated with protein-specific antibodies, and immunocomplexes were collected by incubating the lysate-antibody mix with protein G Dynabeads (Cat# 10004D, ThermoFisher) at 4°C overnight on a rotator. The beads were extensively washed with NP-40 lysis buffer. The immunocomplexes captured by protein G beads were eluted by 1x SDS loading buffer, and the eluted immunocomplex protein content was examined by protein-specific antibodies and IB methods as described above in “Immunoblotting analysis.”
In vivo ubiquitination assay
For in vivo ubiquitination assay, 293T cells were co-transfected with mammalian expression vectors for Flag-tagged STING1 (PCMV3-STING1-Flag), ubiquitin (PCI-His-hUbiquitin), RBX1 (pCDNA3-myc3-ROC1), and Cullin 3 (pcDNA3-myc-CUL3) together with SPOPwt (pcDNA3.1-HA-SPOPwt), SPOPF102C (pcDNA3.1-HA-SPOPF102C), SPOPF133V (pcDNA3.1-HA-SPOPF133V), or pcDNA3.1 vector (control). Thirty-six hours after transfection, PS-341 (250 nM final concentration) was added to the incubation for another 6 hours. The cells were harvested and washed in ice cold PBS and lysed in RIPA buffer. The cell lysates were sonicated and cleared by centrifugation. The supernatants were incubated with PureProteome™ Nickel Magnetic Beads (Cat# LSKMAGH10, MilliporeSigma) to capture ubiquitinated proteins. The beads were extensively washed in RIPA buffer and proteins were eluted by 2x SDS loading buffer containing 500 mM imidazole (Cat# I2399, Sigma-Aldrich). The eluted proteins were resolved by SDS-PAGE for immunoblotting analysis as described above, with anti-Flag antibody to determine the ubiquitination of Flag-tagged STING1 protein. In addition, the RIPA cell lysates from the in vivo ubiquitination experiments were incubated with anti-Flag magnetic beads (Cat# HY-K0207, MedChemExpress LLC, NJ) to capture Flag-tagged STING1 proteins. The beads were extensively washed with RIPA buffer and proteins were eluted by 2x SDS loading buffer. The eluents were examined using a mass spectrometry-based post-translational modifications (PTM) service provided by Poochon Scientific LLC (Frederick, MD) to determine the ubiquitination conjugation motif recognized by SPOP-Cul3 E3 ligase complex.
Colony formation assay
Colony formation assay was used to examine response sensitivity of prostate cancer to PARPi treatment in vitro. Briefly, cells were seeded into 6-well plates at 5,000 cells/well, and drugs were introduced 48 hours after plating. Up to 2 weeks were allowed for colony formation. The cell culture was refreshed every 3 days with fresh culture medium containing the indicated drugs or vehicle control. At the end of the experiments, culture medium was removed and the colonies formed on the plate were fixed by cold methanol and stained with 0.5% crystal violet. The plates were scanned and imaged on a camera-equipped Eclipse TE2000 U microscope (Nikon Instruments, Inc., NY), and the number of colonies was counted using NIS-Elements AR 2.30 software (Nikon Instruments, Inc.). The quantitative results were plotted and displayed as the percentage number of control (vehicle treatment).
MTS cell viability assay
Cells were plated in 96-well plates and treated as indicated for 48 hours. Following the product manual, MTS assay was performed using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Cat# G3581, Promega, WI) and a Biotek Synergy 2 microplate reader (BioTek, CA).
SiRNA transfection
Cells were seeded (in 96- or 6-well plates) one day before siRNA transfection. Following the product manual instruction, the cells were transfected with 20 nM (final concentration) gene-specific siRNA or non-targeting control siRNA (siNC) utilizing the Lipofectamine RNAiMax transfection reagent (Cat# 13778075, ThermoFisher). Forty-eight hours after transfection, the transfected cells were analyzed by MTS, or protein expression as indicated by the designated experiments, following the protocols described above in “Immunoblotting analysis” and “MTS cell viability assay.” The sequences of gene-specific siRNA and siNC are listed in Supplementary Table S5.
Xenograft tumor models
Aliquots of 5 × 106 human C4–2b-pLenti-HA-SPOPF133V or C4–2b-pLenti-EV (SPOPwt control) prostate cancer cells in 100 μL (1:1, PBS:Matrigel®) were subcutaneously injected into the right flanks of 5-week-old male SCID mice (Charles River Lab, Wilmington, MA). Tumors were allowed to grow until they reached 60 to 70 mm3 before they were randomly distributed to receive one of the following treatments orally for 18 days: talazoparib (0.33 mg/kg 5 days/week) or vehicle control buffer [10% dimethyacetamide (Sigma), 6% solutol HS (Sigma), 84% PBS]. Tumor volume was measured twice/week using calipers and calculated on the basis of (width x width x length)/2, as previously described (13,23,24). Eighteen days after the initial treatment, the tumors were harvested. All animal experiments were conducted in accordance with accepted standards of humane animal care approved by the MDACC Institutional Animal Care and Use Committee.
Statistical analysis
Data from at least 3 biological repeats were presented as the mean ± SEM. Two-tailed t tests were used to compare and determine the statistical significance of two individual treatments or combinations in RT-qPCR analyses, colony formation assay, and MTS assay. Two way or three way ANOVA was used for analysis of the significance of tumor growth profiles between each model line and treatment groups. P values <0.05 were considered statistically significant.
Data availability statement
RNA-seq data for RM-1-BM cell lines and RAW264.7 cells used in coculture model experiments were deposited to The Gene Expression Omnibus (GSE240199).
RESULTS
SPOPmut prostate cancer demonstrates increased STING signaling and NF-κB pathway gene expression
Gene set enrichment analysis (GSEA) of an annotated dataset of CRPC patient tumors (dbGap Accession: phs000909.v1p1) revealed significant enrichment of TNF-α-NF-κB signaling genes in SPOPmut [versus SPOP–wild-type (SPOPwt)] CRPC (Fig. 1A, Supplementary Methods). Further expression analysis of the CRPC dataset using a 259-gene set comprising a comprehensive list of canonical cGAS-STING-TBK1 and NC-STING-NF-κB signaling genes (Supplementary Methods and Supplementary Table S1) identified 35 significantly differentially regulated genes, including 29 upregulated genes in SPOPmut CRPC compared to SPOPwt (Fig. 1B). Importantly, all significant upregulated genes were derived from gene sets comprised, in part, from the NC-STING-NF-κB signaling genes included in the 259-gene set used in the analysis, showing the dominance of NC-STING-NF-κB activity (NC-STING) over canonical STING-IFN-β in SPOPmut CRPC (Supplementary Table S1).
Fig. 1. Upregulation of TNF-α-STING-NF-κB and non-canonical STING-NF-κB gene expression in SPOP mutant prostate cancer patients.
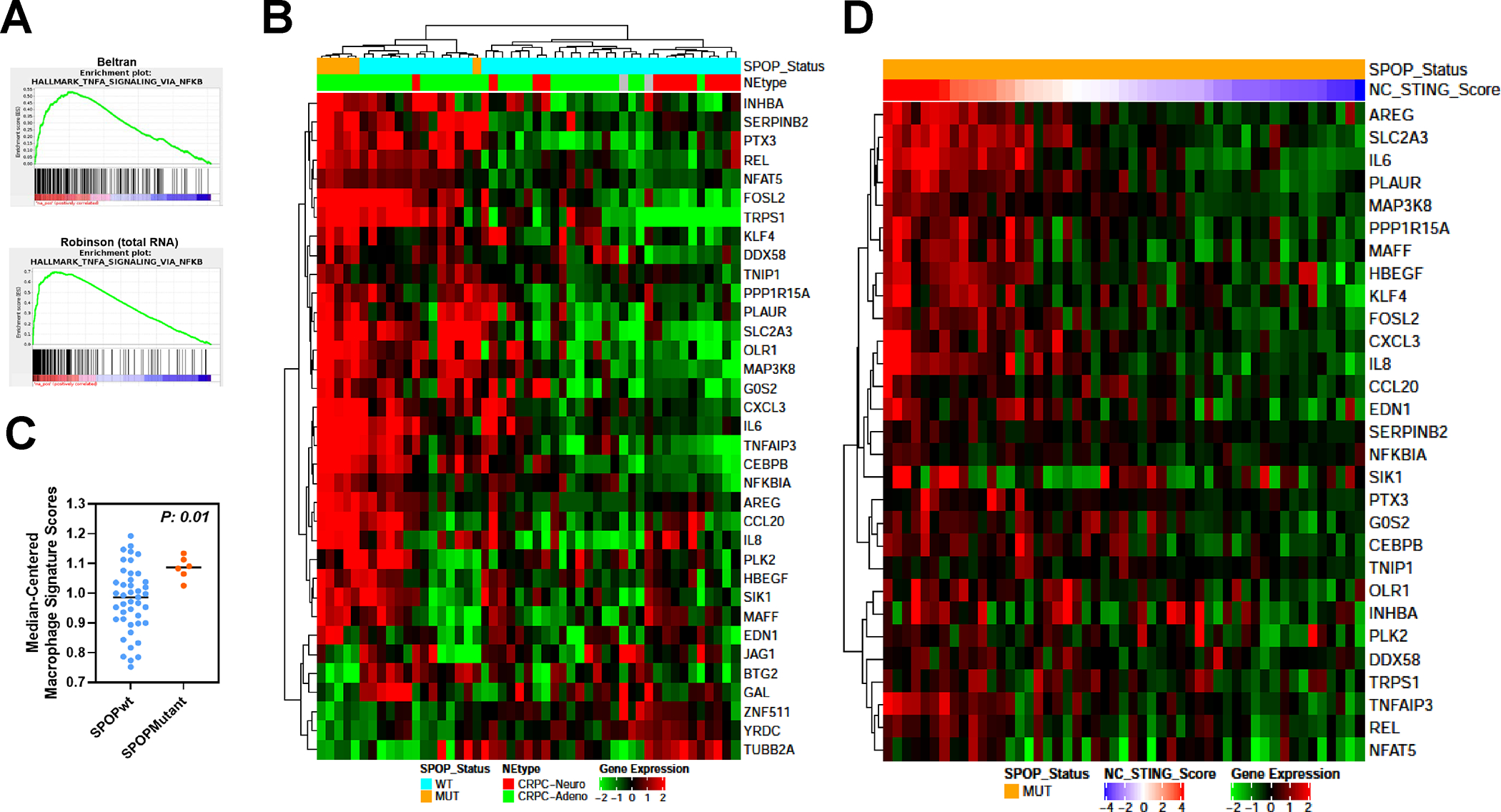
A. GSEA analysis of castration-resistant prostate cancer data sets (Beltran, CRPC-Adeno and CRPC-Neuro; Robinson-total RNA; Robinson- polyA RNA) with a curated gene set (Hallmark) shows enrichment of TNF-α-NFκB signaling genes. B. Heatmap of significantly differentially regulated genes from unsupervised analysis of the same data set using a 259 gene set comprised of canonical cGAS-STING-TBK1 and NF-κB signaling genes identified a cluster of differentially expressed genes in SPOPmut prostate cancer patients compared to SPOP wild-type patients. C. “Immunome” analysis of Beltran CRPC cohort using a compendium of publicly available data from purified immune subsets. D. Heatmap of significantly upregulated genes in SPOPmut prostate cancer from Beltran dataset in B (NC-STING signature genes) using TCGA SPOPmut data set ordered by the z-score of NC-STING signature (NC-STING Score).
We next analyzed the intratumoral “immunome” of this CRPC patient cohort using a compendium of publicly available data from purified immune subsets as previously described (25). Interestingly, macrophages, neutrophils, and memory T cells were enriched in the SPOPmut TME compared to the SPOPwt TME, including a moderately significant increase in M1-like macrophages compared to M2-like macrophages (Fig. 1C and Supplementary Fig. S1) (26). These results are consistent with SPOP mutant CRPC resulting in upregulation of NC-STING activity and an immunosuppressive TME, dominated by macrophages and neutrophils, amongst other immune cell populations.
Lastly, we evaluated for enrichment of the NC-STING signature by developing an “NC-STING Score” based on aggregation of SPOPmut-upregulated signature genes and analyzing primary, hormone-naïve prostate cancer from TCGA (Supplementary Methods). The results of supervised clustering analysis identified a subset of SPOPmut, castrate-sensitive primary prostate cancers that were enriched for the same NC-STING-NF-κB pathway as the androgen-indifferent SPOP mutants from the aforementioned CRPC dataset (Fig. 1D).
Stably transduced SPOPF102C and SPOPF133V prostate cancer cell models demonstrate increased STING signaling
We conducted shotgun proteomics analyses of doxycycline (Dox)-inducible SPOPwt and SPOPmut prostate cancer models to evaluate the changes in the proteome attributed to SPOPmut prostate cancer cells. These analyses yielded 81 differentially expressed proteins between SPOPmut (F102C and F133V) C4–2b prostate cancer models compared to SPOPwt or vector control (VC) (Fig. 2A, upper panel). Ingenuity Pathway Analyses of these 81 proteins revealed enriched representation of an NF-κB–centric protein network (Fig. 2A, lower panel). We generated and analyzed lentivirus-transduced human C4–2b and mouse RM-1-BM CRPC cells that stably express: 1) SPOPwt, 2) SPOPF102C, and 3) SPOPF133V, versus empty lentiviral vector–transduced control cell lines [empty vector (EV) or VC] (Fig. 2B, C and Supplementary Fig. S2). Analysis of these cell lines by immunoblotting (IB) showed upregulation of PARP1, ATM and selected NC-STING-NF-κB-IL-6 signaling pathway proteins HMG proteins that are involved in promoting secretory pathway activities including extracellular vesicle production, compared to VC or SPOPwt isogenic lines (Fig. 2B, C, Supplementary Fig. S2 and Supplementary Tables S6, S7). Notably, upregulation of PARP1 and ATM is consistent with the previously recognized DNA repair activities of SPOP (5). In addition, PARP1 and ATM are critical components of an alternative STING signaling complex involved in the activation of the transcription factor NF-κB and induction of an alternative STING-dependent gene expression program (14). This upregulation was particularly notable in human and mouse prostate cancer-SPOPF133V lines (Fig. 2B, C and Supplementary Fig. S2).
Fig. 2. Upregulation of non-canonical STING-NF-κB/STAT3, canonical cGAS-STING and activation STAT3-HMG secretory pathways in SPOP mutant (SPOPF102C/SPOPF133V)-expressing prostate cancer cells.
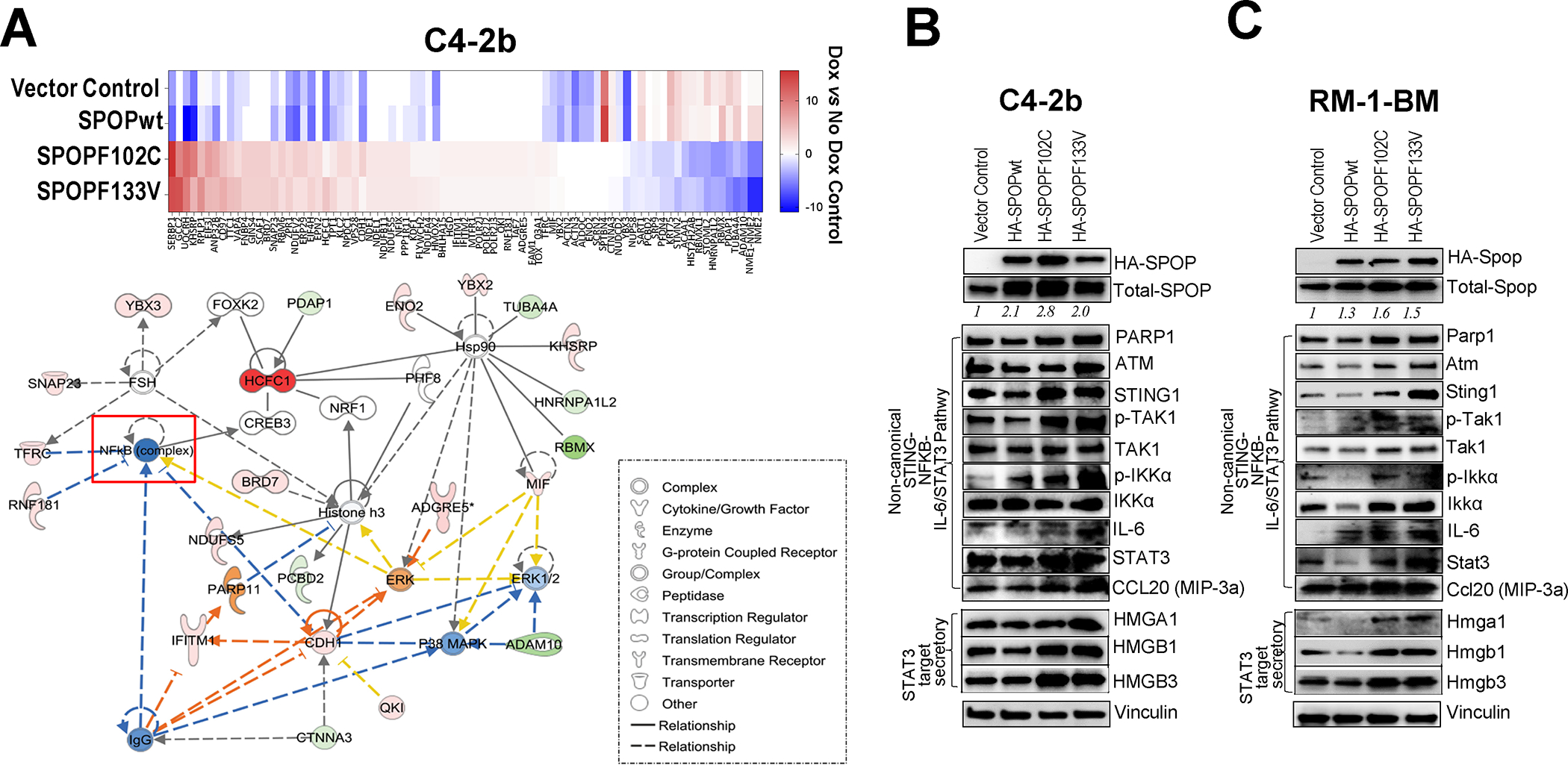
A. Targeted proteomics analysis of Dox-inducible SPOPwt and SPOPmut C4–2b and RM-1-BM prostate cancer models yielded 81 protein features that were differentially expressed between SPOPmut (F102C and F133V) C4–2b prostate cancer cells compared to SPOPwt or empty vector controls (upper panel). Ingenuity Pathway Analyses of these 81 proteins revealed enriched representation of NF-κB-centric protein network features (lower panel). B, C. Immunoblot analysis of proteins involved in non-canonical STING-NF-κB signaling, cGAS-STING signaling and STAT3-HMG secretory regulation pathways in stably transduced SPOPwt- and SPOPmut-(F102C, F133V) expressing or empty vector control C4–2b and RM-1-BM prostate cancer cells.
SPOP mutants mediate stabilization of STING signaling proteins in prostate cancer models
Based on the alignment of published SPOP substrates for SBC motifs, we searched SPOP binding sites in human and mouse non-canonical and canonical STING pathway signaling proteins (4,27). Among the proteins that contained putative SBCs was STING, suggesting that STING1 is an SPOP binding candidate potentially degraded by the E3 ubiquitin ligase activity of the SPOP-Cul3 complex. To test this, we established human (C4–2b) and mouse (RM-1-BM) prostate cancer cell lines that conditionally express Dox-induced SPOPwt or SPOP mutants. Our dose-response characterization studies demonstrated that SPOPmut expression (inducing less than 5–10x compared to controls with no Dox induction) was achieved using 10 to 20 ng/mL Dox. Overexpression of SPOP was achieved using 200 ng/mL Dox. Significantly, the predicted SPOP substrate STING protein was decreased by induced SPOPwt expression, but upregulated by SPOPF102C or SPOPF133V in both cell lines (Fig. 3A and Supplementary Fig. S3). Proteasome inhibitor (PS341) rescued STING1 destabilization in Dox-induced or stably transfected SPOPwt prostate cancer models, suggesting SPOP-dependent ubiquitination and proteasomal degradation of STING1 protein (Fig. 3B, Supplementary Fig. S4 and Supplementary Table S8). Co-IP analysis demonstrated protein–protein interactions of SPOP and STING1 (Fig. 3C and Supplementary Fig. S5). Utilizing 293T cell-expressed SPOPwt and SPOPF102C/SPOPF133V (substrate-binding deficient) as the “prey” and purified STING protein as the “bait”, far western blot assay suggested direct interaction of STING protein with only SPOPwt, which contains an intact substrate-binding motif (Supplementary Figs. S6, S7). Furthermore, in vivo ubiquitination analysis showed that, while co-transfection of SPOPwt increases the ubiquitination of STING1, overexpression of SPOPF102C and especially of SPOPF133V effectively inhibits the ubiquitination of STING1 in 293T cells (Fig. 3D and Supplementary Fig. S8). MALDI-TOF mass spectrometry analysis of ubiquitinated STING1 protein from in vivo ubiquitination samples co-transfected with SPOPwt identified SPOPwt-dependent (i.e., substantially reduced in SPOPF133V co-transfected samples) ubiquitination conjugation site K347 of STING1 protein (Supplementary Fig. S9). In addition, 293T cells were co-transfected with expression vectors to generate constitutive levels of STING1 protein and increased amounts of SPOPwt, SPOPF102C, or SPOPF133V. The results showed that co-expression of SPOPwt, but not SPOPF102C or SPOPF133V, destabilizes and promotes degradation of STING1 in a dose-dependent manner (Fig. 3E and Supplementary Fig. S10). Cyclohexamide (CHX) chase protein half-life assays confirmed the substantially reduced half-life of STING1 by SPOPwt co-expression and increased half-life of STING1 by SPOPF102C or SPOPF133V co-expression in 293T cells (Fig. 3F and Supplementary Fig. S11). Together, these data suggest that STING1 is a bona fide substrate of SPOP and that expression of substrate-binding–deficient prostate cancer–associated SPOP mutants effectively stabilize and elevate the expression of STING1 protein in prostate cancer.
Fig. 3. STING1 is a putative SPOP-interacting substrate and is functionally regulated by expression of substrate-binding deficient SPOP mutation (SPOPF102C, SPOPF133V).
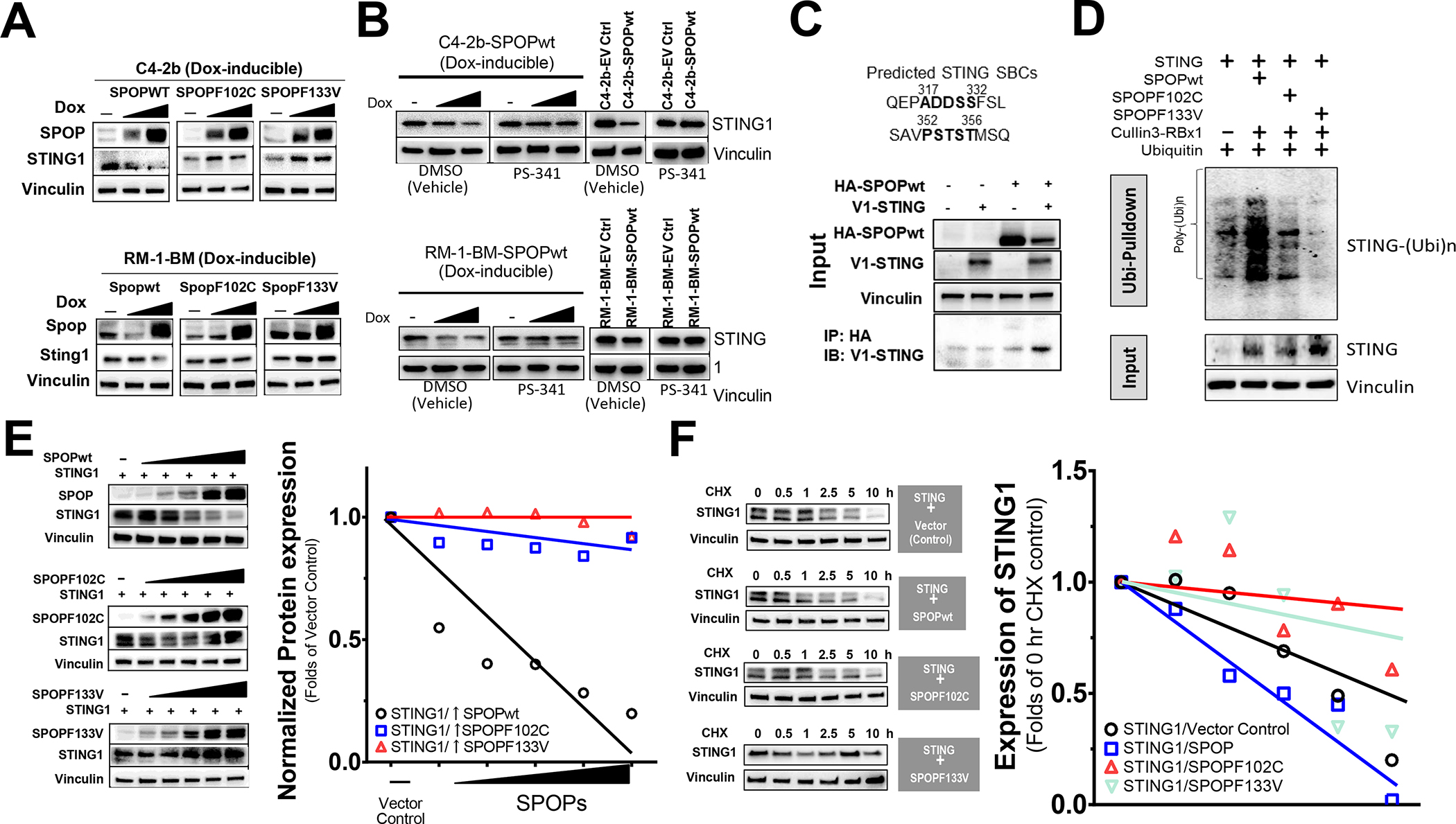
A. Analysis of STING1 protein following induced expression of SPOPwt, SPOPF102C or SPOPF133V in doxycycline-inducible C4–2b and RM-1-BM cells (induced with the dose of doxycycline at 0, 10 or 200 for C4–2b-SPOPs and 0, 20 or 200 for RM-1-BM-SPOPs, respectively). B. Analysis of STING1 protein in proteosome inhibitor (PS-341) treated, Dox-induced and stably transfected SPOPwt models. C. Co-IP analysis of protein-protein interactions of SPOP and STING1. D. In vivo ubiquitination assay to examine the polyubiquitination of STING1 protein by SPOP E3 ligase complex (SPOP-Culin3- RBX1) or SPOPmuts (SPOPF102C, SPOPF133V) cotranfected in 293T cells. E. Analysis of SPOPwt and STING1 following cotransfection in 293T cells. F. Cycloheximide (CHX) chase protein half-life assay of STING1 following SPOPwt, SPOPF102C or SPOPF133V co-expression.
SPOP mutants potentiate PARPi-induced growth inhibition through enhanced induction of cGAS-STING and suppression of NC-STING- NF-κB signaling in prostate cancer models
Based on a previous publication showing that SPOPmut prostate cancer cells are more sensitive to PARPis than SPOPwt prostate cancer cells in vitro (5), as well as our own analysis demonstrating that STING1 is a bona-fide SPOP target and that SPOPmut models manifest a dominant NC-STING signaling phenotype, we tested the effects of STING1 siRNA (siSTING) on response to PARPi olaparib (OLA) in stably transduced SPOPmut prostate cancer models and SPOPwt VCs. Analysis of siSTING- and siNC-transfected, stably transduced human (upper panel) and mouse (lower panel) SPOPmut models confirmed enhanced growth suppression in SPOPmut prostate cancer cells compared to VCs (OLA did not substantially suppress RM-1-BM SPOPwt VCs). STING1 siRNA similarly suppressed growth in VC and SPOPmut prostate cancer cells compared to siNC in the absence and presence of OLA and enhanced OLA-mediated growth suppression (Fig. 4A and Supplementary Figs. S12, S13). To further analyze the effects of PARPi on cell growth—independent of selection for SPOPmut-altered phenotypic properties—we treated Dox-inducible, dose-regulated expression–controlled, SPOPF102C- or SPOPF133V-expressing C4–2b and RM-1-BM prostate cancer models with OLA. Colony formation (Fig. 4B) and MTS (Fig. 4C) assays using these models showed significantly enhanced inhibition of colony formation and cell proliferation in SPOPmut (F102C or F133V)–expressing C4–2b and RM-1-BM cells treated with OLA (MTS) (Fig. 4C). IB analysis of these cells showed that, compared to VCs (SPOPwt expression), Dox-induced, dose-dependent SPOPmut expression enhanced OLA-induced DNA damage and cGAS-STING signaling pathway activities (phosphorylation of STING1 and IRF3), resulting in enhanced IFN-β protein expression (Fig. 4D, Supplementary Figs. S14, S15 and Supplementary Tables S9, S10). This response is consistent with DDRi-induced, cGAS-STING-IFN-β–mediated growth suppression for prostate cancer cells (13) and with enhanced OLA-induced growth inhibitory activity in Dox-inducible human (C4–2b) and mouse (RM-1-BM) prostate cancer models (Fig. 4B and 4C, respectively) and stably transduced, SPOPmut-expressing prostate cancer models (Fig. 4A). Taken together, the results of these experiments indicate that SPOPmut-mediated STING stabilization allows for OLA-driven maintenance of canonical c-GAS-STING signaling, which can override the dominant NC-STING-NF-κB survival signaling in SPOPmut prostate cancer.
Fig. 4. SPOP mutants potentiate growth inhibition and induction of cGAS-STING by PARP inhibitor treatment, associated with increased inhibitory STAT3 phosphorylation and suppression of secretory signaling targets and non-canonical STING-NF-κB signaling in prostate cancer models.
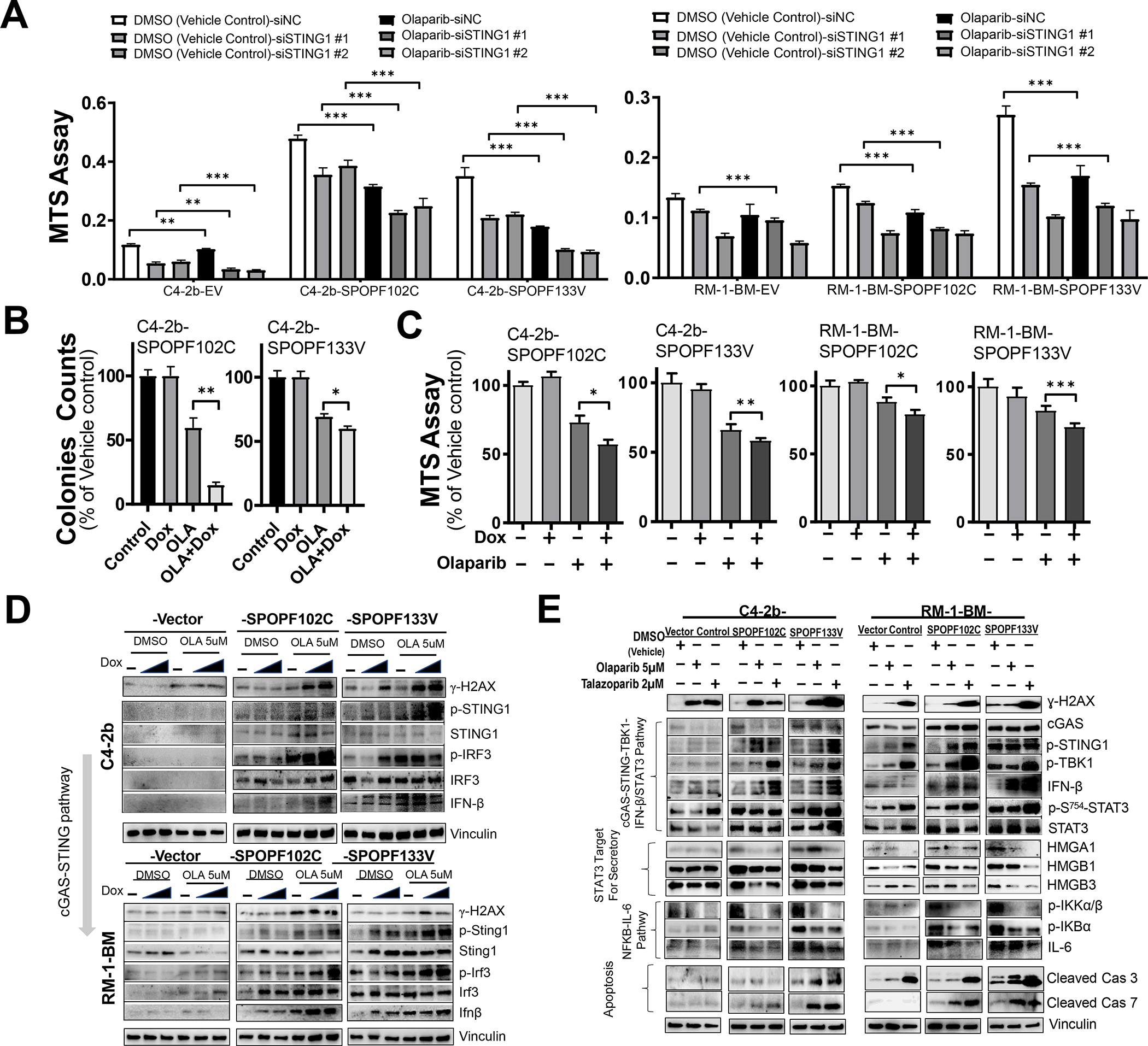
A. MTS analysis of C4–2b and RM-1-BM (SPOPF102C or SPOPF133V) treated with siSTING or siNC in the presence and absence of olaparib (OLA), *, P<0.05, and **, P<0.01. B. Colony formation and C. MTS analysis of cell proliferation in doxycycline (Dox)-induced SPOP mutant-expressing C4–2b and RM-1-BM (SPOPF102C or SPOPF133V) in the presence and absence of OLA. T-test was used for statistical analysis, *, P<0.05, and **, P<0.01. D. Immunoblot analysis of OLA-induced DNA damage and canonical STING1 signaling activities (phosphorylation of STING1 and IRF3, and IFN-β) in Dox-induced C4–2b and RM-1-BM SPOP mutants compared to empty vector controls in a dose-dependent manner (Dox 10 and 200 ng/mL for C4–2b-SPOP models, or 20 and 200 ng/mL for RM-1-BM-SPOP models). E. Analysis of the effects of PARP inhibitor (OLA and talazoparib, TALA) on DNA damage (γH2AX), cGAS-STING-TBK1 signaling, IFN-β protein expression, p-S754-STAT3 expression, HMGA1/HMGBs secretory signaling protein expression, NF-κB signaling, IL-6 expression and expression of proapoptotic signaling proteins (cleaved caspases 3 and 7) in human and mouse SPOPmut models compared to empty vector control cells.
To define the molecular mechanism by which PARPi leads to inhibition of NC-STING-NF-κB-IL-6/STAT3 signaling and induction of canonical cGAS-STING-IFN-β in prostate cancer epithelial cells, we treated SPOPmut (F102C, F133V) and VC C4–2b and RM-1-BM cells with OLA, talazoparib (TALA), or vehicle control. As expected, PARPi (OLA and talazoparib, TALA) resulted in increased DNA damage (γH2AX), and SPOPmut (F102C and F133V) expression in prostate cancer cells led to enhanced activation of STING1 (p-S366-STING1) (Fig. 4E, Supplementary Figs. S16, S17 and Supplementary Tables S11, S12) (28). Notably, the differential responses in SPOPmut compared to VC models following OLA or TALA were similar. Interestingly, SPOPF133V showed increased cGAS-STING activation compared to SPOPF102C, which correlated with increased induction of DNA damage following PARPi treatment (Fig. 4E). Notably, PARPi led to cleavage of caspase (Cas) 3, which was enhanced in both human and mouse SPOPF133V prostate cancer models, suggesting that these PARPis induce Cas3 cleavage–dependent cell death. More importantly, increased cleavage of Cas7 was observed only in the SPOPF102C or SPOPF133V prostate cancer models in which enhanced cGAS-STING-TBK1 signaling and substantially increased expression of IFN-β was observed following PARPi treatment, and not in the prostate cancer models without SPOPmut expression (VC) (Fig. 4E). While IFN-β–induced cell death reportedly depends on Cas7 cleavage (29), these observations suggest the involvement of PARPi-activated cGAS-STING1-TBK1-IFN-β-Cas7, in addition to PARPi-induced Cas3–dependent cell death, as an important mechanism contributing to the enhanced cell death/growth inhibition of the SPOPmut prostate cancer models. Autocrine and paracrine positive feedback upregulation of the IL6-STAT3 pathway and elevated activation of STAT3 are critical for the growth of many advanced cancers, including CRPC (30,31). We examined the potential link between cGAS-STING1-TBK1 and STAT3 activation through IB analysis of TBK1-dependent STAT3-inhibitory phosphorylation (p-S754-STAT3) (32). The results demonstrated markedly increased p-S754-STAT3 in PARPi-treated human and mouse SPOPmut models compared to control cells (Fig. 4E). Inhibition of STAT3 by TBK-mediated S754 phosphorylation can suppress transcriptional activation and secretion of IL6, which in turn could translate into suppression of adaptive feedback resistance signaling pathways based on IL6 autocrine and paracrine mechanisms (33). This mechanistic concept is supported by the results of additional IB analysis demonstrating PARPi-mediated downregulation of p-IKKα/ß and p-IKBα (Fig. 4E).
SPOP mutants promote PARPi-induced macrophage reprogramming through soluble factors
As shown in Figure 1 and Supplementary Fig. S1, macrophages are enriched in SPOPmut CRPC. Thus, to analyze the effects of PARPi in monoculture compared to coculture conditions, we used a transwell coculture system (tumor cells in upper chamber, macrophages in lower chamber). The results of these experiments showed that while expression of SPOP mutants (F102C or F133V) sensitizes both C4–2b and RM-1-BM prostate cancer cells to OLA, when cocultured with differentiated (phorbol-12-myristate-13-acetate, PMA) THP-1 (for C4–2b) or RAW264.7 (for RM-1-BM) macrophages, OLA-induced growth inhibition was enhanced [~-20% (C4–2b) and ~-40% (RM-1-BM)] compared to VCs (Fig. 5A, B, respectively). OLA-induced growth suppression was partially inhibited by incubation with anti-IFN-β neutralization antibodies and restored by anti-IL-6 neutralization antibodies; these effects were more pronounced in SPOPF133V-expressing C4–2b and RM-1-BM prostate cancer cells (compared to SPOPF102C cells) (Fig. 5A, B). Importantly, OLA combined with GW4869, a neutral sphingomyelinase inhibitor that blocks exosome generation and secretion, abrogated these SPOPmut-sensitized responses of macrophage-cocultured prostate cancer cells to OLA treatment (Fig. 5A, B). In addition, quantitative real-time PCR (RT-qPCR) (Fig. 5C, D) and IB (Fig. 5E, Supplementary Fig. S18 and Supplementary Table S13) analyses demonstrated marked activation of cGAS-STING signaling, including IFN-β production, selectively in the macrophages (THP-1 or RAW264.7) cocultured with OLA-treated SPOPmut (F102C or F133V) C4–2b or RM-1-BM cells, but not in monocultured macrophages or macrophages cocultured with SPOPwt VC C4–2b or RM-1-BM cells (Fig. 5C, D, E). Taken together, these results suggest that OLA treatment directs SPOPmut prostate cancer cell–mediated phenotypic reprogramming, leading to induction of cGAS-STING-IFN-β signaling in tumor-associated macrophages (TAMs) through SPOPmut-activated secretory activity, such as soluble immunoregulatory protein effectors (34). We also showed that PARPis induced TBK1-dependent STAT3-inhibitory phosphorylation (p-S754-STAT3), and downregulation of STAT3-regulated HMG proteins that are involved in promoting secretory pathway activities (Figs. 2B, C and 4E). To show interaction between SPOPF133V genotype and PARP inhibition we used bulk RNA-sequencing analysis of RM-1-BM-SPOPF133V versus empty vector control cells, which were monocultured or cocultured with RAW264.7 macrophages and treated with OLA or control DMSO. The results depicted on heatmaps from RM-1-BM-SPOPF133V and RAW264.7 macrophages cocultured cells revealed 42 common differentially expressed genes demonstrating significant interaction between expression of the SPOP mutation and OLA treatment in cocultured RM-1-BM-SPOPF133V mutant–expressing prostate cancer cells and RAW264.7 cells (Fig. 5F), and 11 common differentially expressed genes demonstrating significant interaction between expression of the SPOP mutation and OLA treatment in cocultured and monocultured RM-1-BM-SPOPF133V mutant–expressing prostate cancer cells (Supplementary Fig. S19). These data reveal SPOPmut-driven, concurrent changes in gene expression in prostate cancer epithelial cells and macrophages, that likely contribute functionally to an SPOPmut prostate cancer phenotype in this model. Importantly, OLA treatment substantially altered these gene expressions and appears to mitigate many of the gene activities in both cell types. Notably, one of the upregulated SPOPmut and PARPi interactive genes, i.e., SGK1 corresponds to a pathway previously shown to represent potentially targetable SGK1-regulated pathways that contribute to human prostate cancer cell survival and potential drug resistance in response to PARP inhibition (35). In addition, multiple interactive genes were identified that were responsive to TNF-α (Tnfrsf9), (36) and IFN-ß (Irf7 and Ifih1) (37,38), which is consistent with the paracrine role of these factors demonstrated in our coculture experiments (Fig. 5 and Supplementary Table S13). Thus, given the critical roles of IL-6 in modulating macrophage polarization and regulating differentiation of other immune cells including T cells (39,40), these findings point to the possibility that PARPi treatment of SPOPmut prostate cancer may functionally modulate the TME through alteration of non-canonical to canonical STING signaling to promote therapeutic efficacy in vivo.
Fig. 5. Expression of SPOP mutations in prostate cancer cells enhance PARP inhibitor-mediated growth inhibition through paracrine activities in prostate cancer-macrophage coculture models.
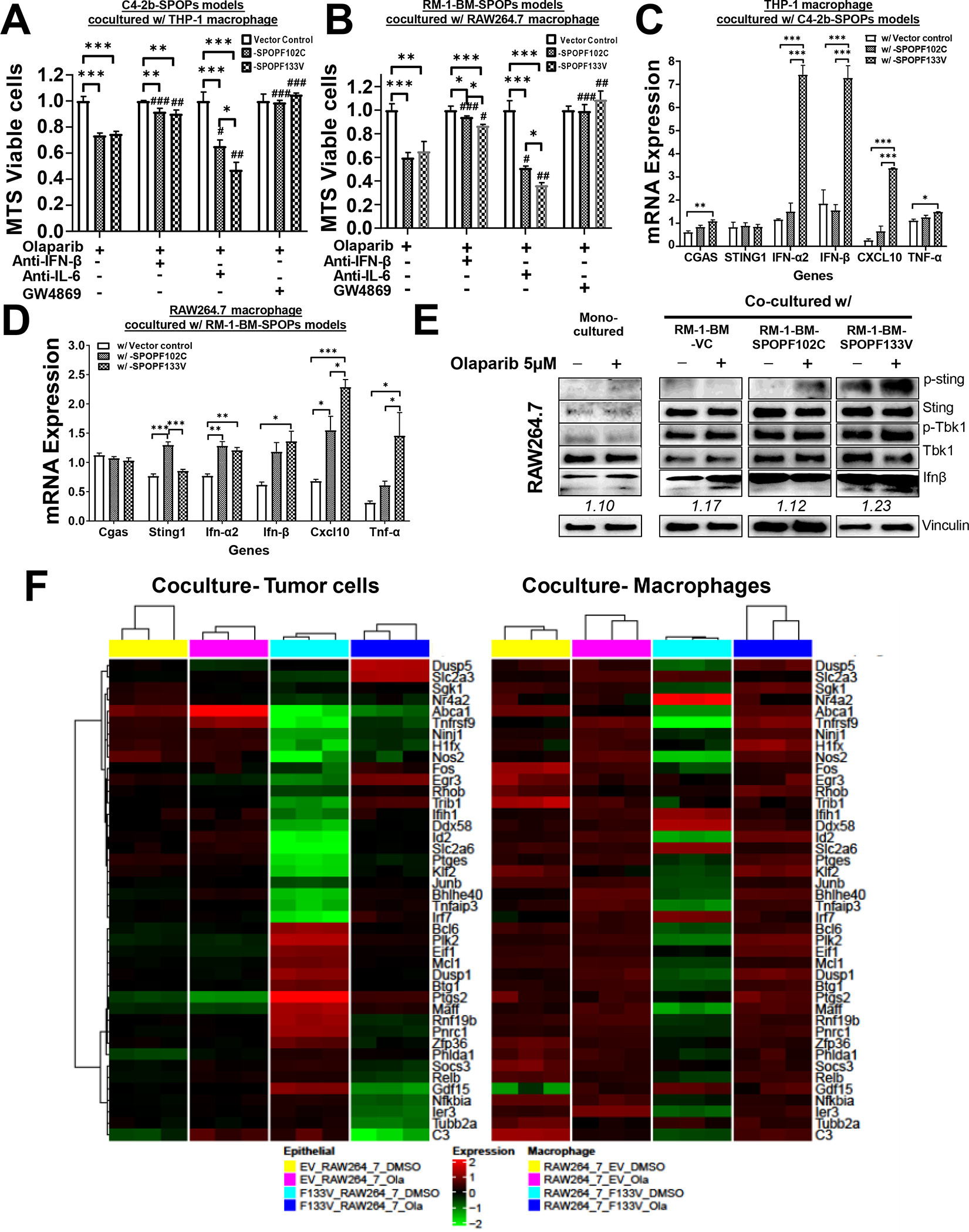
A, B. Expression of SPOP mutant (F102C or F133V) sensitized C4–2b and RM-1-BM prostate cancer models to OLA-mediated growth inhibition. Data were normalized to monocultures of the designated cells following DMSO (control) and OLA treatment. C, D. RT-qPCR analysis of cGAS-STING signaling and IFN-β production specifically in macrophages (THP-1 or RAW264.7) that were cocultured with OLA-treated C4–2b and RM-1-BM cells expressing SPOP mutants (SPOPF102C or SPOPF133V) or vector controls. E. IB analysis of p-Sting-p-Tbk1-IFN-β signaling in RAW264.7 cells in monoculture and coculture [with RM-1-BM prostate cancer models expressing SPOPmuts (SPOPF102C and SPOPF133V) or empty Vector Control (VC)]. A-D. * One-way ANOVA was used to compare the differences in one group, #T test for compare the differences between OLA and other OLA combination treatments, respectively. *or # P<0.05, **or ## P<0.01, ***or ### P<0.005. Bar legend for panels A-D is shown in panel C. F. Heatmaps of common differentially expressed genes in cocultured RM-1-BM-SPOPF133V prostate cancer cells and macrophages to show significant (FDR < 0.05) interaction between treatment [OLA or vehicle control (DMSO)] and SPOP phenotype.
SPOP mutation (F133V) enhances PARPi-induced growth suppression in human prostate cancer xenograft models
To test the efficacy of PARP inhibition in suppressing the growth of human prostate cancer cells in vivo within the context of SPOP mutation, we generated SPOPF133V and VC C4–2b xenograft models and treated them with TALA or vehicle control. The results showed greater PARPi-induced growth suppression in SPOPF133 tumor compared to VC tumor (Fig. 6A, B). Immunostaining analysis from representative tumor samples demonstrated substantially increased p-STING1 (Fig. 6C) and p-S754-STAT3 (Fig. 6D) selectively in SPOPF133V tumors following TALA treatment. These results indicate enhancement of activation of cGAS-STING signaling in SPOPF133V C4–2b tumor cells compared to C4–2b VC control tumor cells and are in agreement with the results of IB analysis (Fig. 4E). Further analysis of SPOPF133V and VC C4–2b xenograft tumors using immunofluorescence staining revealed marked alterations in PARP1-associated nuclear size and PARP1 intracellular distribution in SPOPF133V-expressing C4–2b tumor cells compared to C4–2b VC tumor cells (Supplementary Fig. S20). In addition, double-labeling immunofluorescence staining revealed associations of PARP1 and cleaved Cas3 in association with the appearance of micronuclei in SPOPF133V-expressing tumor cells compared to C4–2b VC control tumor cells (Supplementary Fig. S21). Activation of p-STING1 and immune response may be enhanced as well in part due to a greater degree of dying cells in the SPOPmutant cancer treated with PARPi. Consistent with these observation, immunfluorescence staining demonstrated significantly higher DNA damage (indicated by γ-H2AX), IFN-β expression and cleaved caspase 7 in C4–2b-SPOPF133V prostate cancer tumors compared to VC control tumors in vivo (Fig. 6E, F and G). Importantly, while cleavage of caspase 7 has been reported as the proapoptotic markers by cytotoxic activity of IFN-β (13,29) these observation suggest the significant and functional role of IFN-β that was potentially from interaction of tumor-host macrophages during the TALA treatment in vivo, to mediate enhanced growth inhibition of C4–2b SPOPF133V tumor compared to VC control (Fig. 6A, B).
Fig. 6. SPOPmut prostate cancer demonstrates enhanced sensitivity to PARP inhibitor therapy.
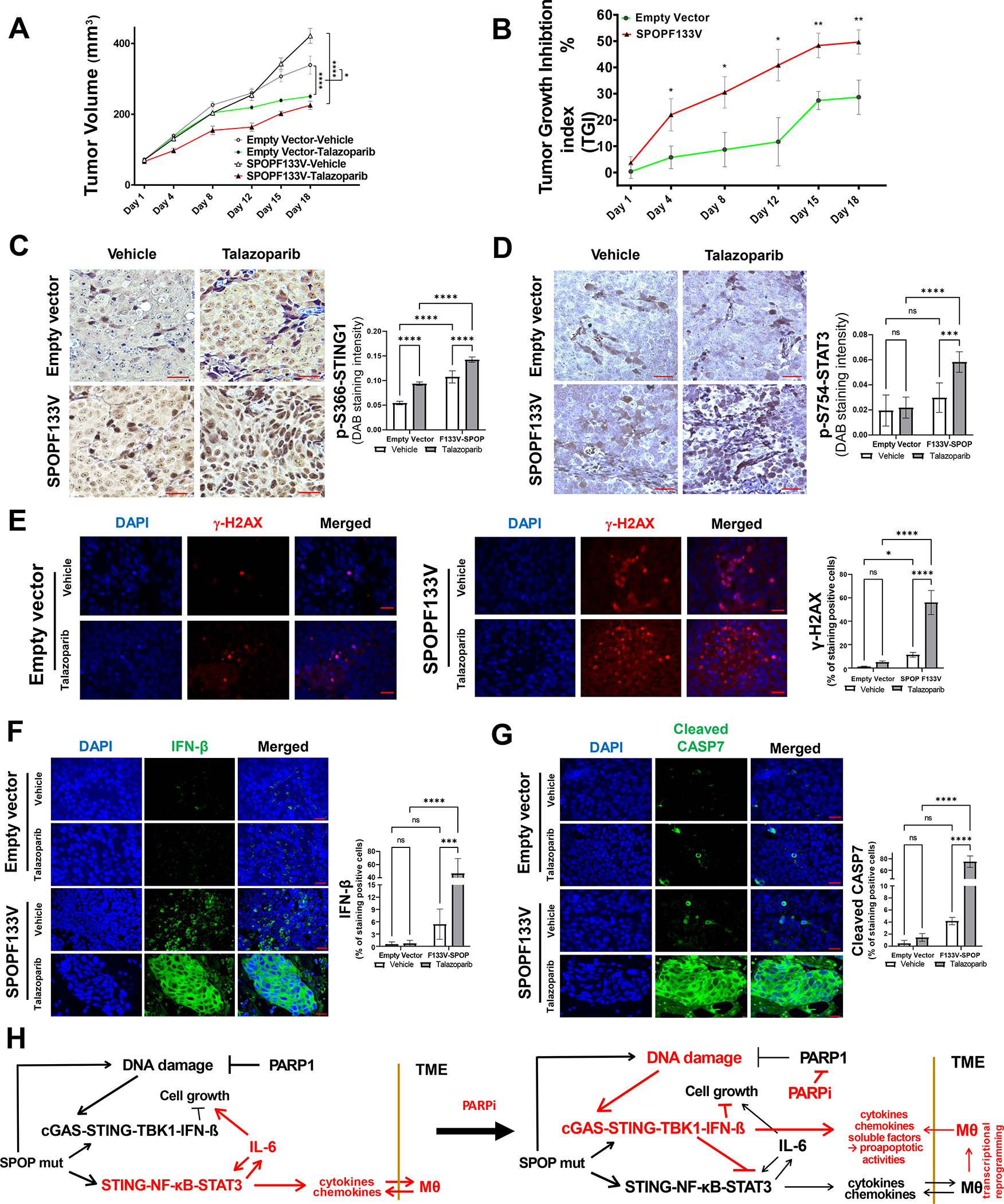
A. Tumor growth during vehicle or talazoparib (TALA) treatment in C4–2b vector controls and C4–2b-SPOP-F133V mutant expressing xenograft models. Two-way ANOVA was used to analyze the statistical significance of control vehicle vs TALA-treated C4–2b-empty vector tumor growth or C4–2b-SPOPF133V tumor growth, respectively, ****, P<0.0001. In addition, three-way ANOVA test was used to analyze TALA treatment affected growth of C4–2b-SPOPF133V tumors (−/+TALA) vs C4–2b-empty vector tumors (−/+TALA), *, P=0.0105. B. Tumor growth inhibition (TGI) index analysis of talazoparib-induced growth suppression in C4–2b-SPOP-F133V mutant expressing xenograft models compared to vector controls. For each individual day point, t-test is used to determine the significance of C4–2b-SPOPF133V vs C4–2b-empty vector tumor TGI %, **, P<0.01 and *, P<0.05. C, D. Immunostaining analysis shows significantly increased p-S366-STING1 and inhibitory p-S754-STAT3 in tumors from A and B, respectively (Bar inset=200 μm). E, F, G. Immunofluorescence (IF) analysis to examine ɣ-H2AX (E), Interferon-β (F), and cleaved caspase 7 (G) in tumors from A and B, respectively (Bar inset=25 μm). Quantitative data of immune staining or IF signals shown in C-G are MEAN ± SD (n=5), ***p<0.001, ****p<0.0005, two-way ANOVA. H. Mechanistic interactions that underlie the “switch” from non-canonical STING signaling to canonical STING signaling shown before and after PARP inhibitor treatment in SPOPmut prostate cancer.
Overall, our results reveal NF-κB-STAT3/IL-6 signaling that is induced and selected for survival in SPOPmut prostate cancer; and this immunosuppressive, pro-survival NC-STING pathway is suppressed by PARP inhibition. Mechanistically, our results show that this suppression is driven in part by SPOPmut–mediated stabilization of STING1, enhanced DNA damage and activation of canonical STING-TBK1 signaling and production of IFNβ, direct inhibition of STAT3 (increased p-S754-STAT3) and activation of proapoptotic pathways (Fig. 6C, D, E, F, G and Supplementary Figs. S20, S21). In addition, the results of SPOPmut and macrophage coculture models and in vivo xenograft experiments indicate that SPOPmut prostate cancer cells can reprogram TAMs that may lead to further suppression of prostate cancer growth (Figs. 5, 6 and Supplementary Fig. S19, S20, S21).
DISCUSSION
We report here that prostate cancer – associated SPOP mutations (in particular the most commonly found variant, F133V) stabilize STING1, leading to activation of both non-canonical and enhanced canonical STING1 signaling in prostate cancer cells. Importantly, analysis of a 259-gene set that contained both non-canonical and canonical STING1 pathway components revealed the non-canonical NC-STING-NF-κB pathway was significantly enriched in SPOPmut, lethal androgen indifferent prostate cancer patients versus SPOP wildtype. The same pattern was demonstrated in models that expressed SPOP mutations (Figs. 1, 2). Deregulated STING1 activities have previously been associated with increased tumorigenesis and poor prognosis (41,42). However, to our knowledge, association of STING stabilization and induction of STING-regulated signaling with a specific DDR-associated mutation, i.e., SPOP, has not been reported. Furthermore, supervised clustering analysis showed a portion of SPOPmut hormone naïve primary prostate cancers were enriched for this immunosuppressive, pro-survival NC-STING pathway (Fig. 1D).
Consistent with previous studies (5), we showed that this SPOPmut phenotype generated a therapeutic vulnerability to PARPis that was associated with increased DNA damage, and in addition, demonstrated a shift from predominant NC-STING-NF-κB signaling to canonical cGAS-STING that promoted growth suppression and proapoptotic signaling (in vitro and in vivo) (Figs. 4–6). Importantly, our results show mechanistically that in addition to DNA damage–associated canonical cGAS-STING-IFN-β induction, PARP inhibition was also associated with TBK1-mediated suppression of STAT3 through induction of p-S754-STAT3 in human and mouse SPOPmut models, which effectively led to inhibition of the STAT3/IL-6 adaptive feedback resistance signaling loop and suppression of NF-κB activation (Figs. 4E, 5, 6 and Supplementary Fig. S19). Previous publications demonstrated that PARP1 is essential and directly interacts, either through or independently of its PARylation enzymatic activity, with NF-κB subunits (p50, p65, or both) to promote NF-κB target gene transcription and production of NF-κB-inducible cytokines (43). Together with our findings, the results of these publications indicate that PARPis regulate STING-dependent and -independent NF-κB activities, and novel biomarkers and therapeutic targets that center on these switches warrant further investigation. More recently, PARP1 was reportedly found in cytoplasm to suppress the cGAS-STING innate immune signaling pathway (“canonical STING” signaling pathway) through PARylating cGAS on D191 (human) or E176 (mouse), inactivating cGAS protein for DNA binding, which depends on DNA damage signaling activation of DNA-PK to phosphorylate T594 on PARP1 for its cytosolic translocation (44). In agreement with our results, these observations provide additional, supportive mechanistic evidence for PARPi-induced, altered NC-STING-NF-κB-IL-6/STAT3 survival signaling to canonical cGAS-STING-TBK1-IRF3-IFN-β signaling in prostate cancer.
The phenotypic alterations associated with CRPC include changes in the TME. It has long been understood that TAMs can either facilitate or antagonize anticancer therapy (45–47). Prostate cancer TAMs can substantially regulate tumor progression, and a recent report showed an increase in CD206-positive macrophages in mCRPC compared to normal prostate (48). Our results demonstrated that PARPi-mediated growth inhibition was, in part, mediated by IFN-ß, whereas IL-6 antagonized these effects in SPOPmut C4–2b and RM-1-BM cells cocultured with macrophages compared to monoculture (Fig. 5A, B). RNA-seq analysis revealed interaction between PARP inhibition and SPOP mutation in reprogrammed RM-1-BM-SPOPF133V cell and macrophage gene expression through soluble factors in RM-1-BM-SPOPF133V and macrophage cocultures. Specifically, our results demonstrated that PARPi interaction with RM-1-BM-SPOPF133V (compared to RM-1-BM empty vector controls) led to regulation of a subset of common differentially expressed genes that were specifically modulated by the coculture conditions (Fig. 5F and Supplementary Fig. S19). These results suggest the development of similar approaches for identification of novel gene mutation-based resistance pathways and therapy targets.
DDR mutations are seen in upwards of 25% of metastatic prostate cancers, with BRCA2 loss serving as the archetype of synthetic lethality with PARPi, though clinical benefit is heterogeneous. Interestingly, a recent publication showed that micronuclei induced by loss of BRCA2 initiate a canonical cGAS-STING response, which involves rewired TNF-α signaling and enhances TNF-α sensitivity (49). Cancers with deleterious mutations in BRCA2 have also been shown to upregulate the NF-κB survival pathway (50). Given this data and our data herein, it is possible that pro-tumorigenic TME alterations resulting from defects in specific DDR genes other than just SPOPmut may support DNA damage–induced cGAS-STING signaling. Our results establish the foundation for application of a novel paradigm for gene-based prognostication and prediction of benefit from PARPi-based therapies in prostate cancer beyond just BRCA gene mutations.
In summary, we show that prostate cancer-associated SPOP mutations stabilize STING1 and effectively lead to predominant activation of pro-tumor, immunosuppressive non-canonical STING1 signaling in CRPC. Non-canonical STING-NF-κB signaling was the predominant pathway that emerged in a gene expression signature derived from SPOPmut CRPC. In addition, a portion of patients with primary, hormone-naïve prostate cancer are enriched for this NC-STING signature; and further studies assessing the prognostic and predictive value of this signature are ongoing in longitudinal, correlative rich clinical trials for patients with castrate sensitive prostate cancer (e.g., NCT04947254). It is possible that this balance favoring non-canonical over canonical STING pathways downstream of DNA damage may in part explain heterogeneity in clinical outcomes seen within DDR mutant subgroups, including SPOP. The ability to identify patients from these heterogeneous subgroups with prostate cancers that are enriched for immunosuppressive non-canonical STING signaling and at greatest risk for developing lethal, androgen indifferent disease will guide biomarker-directed combination treatment strategies such as the use of PARP inhibition and AR signaling inhibition earlier in the treatment course for advanced prostate cancer.
Although the mechanistic studies herein point to a potential paradigm shift in gene-based prognostication and prediction of benefit from PARPi-based therapies in prostate cancer, the results are limited by the use of immunodeficient mice, thus important questions on central immune pathways affected in this context are crucial to answer in subsequent preclinical and clinical studies. In addition, patient-based, biomarker-rich studies that test the interactions between certain gene mutations and PARPis will be necessary to place our results in clinical context. In this regard our ongoing clinical trials for patients with castrate sensitive prostate cancer (NCT04947254) will contribute to this contingency.
Our studies mechanistically link the most prevalent point gene mutations in prostate cancer—SPOP mutations—to NC-STING-NF-κB signaling and show that PARPis can shift NC-STING signaling to anti-tumor canonical cGAS-STING signaling in both the tumor cells and TAMs to enhance the therapeutic activity of this targeted therapy. Further studies into the intersection of STING pathway signaling and PARP1’s role in TME remodeling within the context of the androgen sensitivity spectrum in prostate cancer are warranted, and will be imperative to guide subsequent biomarker-directed combination therapies, including potential immune therapy and DDRi combinations for patients with advanced prostate cancer.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Somatic heterozygous missense mutations in the SPOP substrate-binding cleft are found in up to 15% of prostate cancers. While mutations in SPOP predict for benefit from androgen receptor signaling inhibition (ARSi) therapy, outcomes for patients with SPOP-mutant (SPOPmut) prostate cancer are heterogeneous and targeted treatments for SPOPmut castrate-resistant prostate cancer (CRPC) are lacking. We show in preclinical CRPC models that SPOP targets and destabilizes STING1 protein, and prostate cancer – associated SPOP mutations result in upregulated NC-STING-NF-κB signaling and macrophage- and tumor microenvironment (TME)-facilitated reprogramming, leading to tumor cell growth. We provide mechanism-based evidence that PARP inhibitor treatment results in a shift from immunosuppressive NC-STING-NF-κB signaling to anti-tumor, canonical cGAS-STING-IFN-β signaling in SPOPmut CRPC and results in enhanced tumor growth inhibition. These results define novel underlying mechanisms and clinical markers to guide subsequent biomarker-directed combination therapies for patients with advanced prostate cancer.
ACKNOWLEDGEMENTS
We acknowledge the editorial assistance of Sarah E. Townsend. This research was supported by MD Anderson NCI Prostate Cancer SPORE grant P50 CA140388, R21 CA255581, U01 CA224044, the NCI Cancer Center Support grant P30 CA16672, Weill Cornell Medicine Prostate Cancer SPORE grant P50 CA211024 (to CE Barbieri and M Loda), Department of Defense grants W81XWH2210504_BC211174 and W81XWH-21-1-0282_OC200482 (to T. A. Yap), NCI R01 grant CA255074 (to T. A. Yap) and a Young Investigator’s Award from the Prostate Cancer Foundation (to P.G. Pilié).
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161(5):1215–28 doi 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012;44(6):685–9 doi 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A 2013;110(17):6997–7002 doi 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell 2009;36(1):39–50 doi 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015;4 doi 10.7554/eLife.09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hjorth-Jensen K, Maya-Mendoza A, Dalgaard N, Sigurethsson JO, Bartek J, Iglesias-Gato D, et al. SPOP promotes transcriptional expression of DNA repair and replication factors to prevent replication stress and genomic instability. Nucleic Acids Res 2018;46(18):9484–95 doi 10.1093/nar/gky719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agarwal N, Azad AA, Carles J, Fay AP, Matsubara N, Heinrich D, et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-controlled, phase 3 trial. Lancet 2023. doi 10.1016/S0140-6736(23)01055–3. [DOI] [PubMed] [Google Scholar]
- 8.de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 2020;382(22):2091–102 doi 10.1056/NEJMoa1911440. [DOI] [PubMed] [Google Scholar]
- 9.Pilie PG, George A, Yap TA. Patient selection biomarker strategies for PARP inhibitor therapy. Ann Oncol 2020;31(12):1603–5 doi 10.1016/j.annonc.2020.09.017. [DOI] [PubMed] [Google Scholar]
- 10.Swami U, Isaacsson Velho P, Nussenzveig R, Chipman J, Sacristan Santos V, Erickson S, et al. Association of SPOP Mutations with Outcomes in Men with De Novo Metastatic Castration-sensitive Prostate Cancer. Eur Urol 2020;78(5):652–6 doi 10.1016/j.eururo.2020.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 2019;9(5):646–61 doi 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 2019;79(2):311–9 doi 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Z, Pilie PG, Geng C, Manyam GC, Yang G, Park S, et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin Cancer Res 2021;27(17):4898–909 doi 10.1158/1078-0432.CCR-21-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol Cell 2018;71(5):745–60 e5 doi 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunsch C, Lang RK, Rosen CA, Shannon MF. Synergistic transcriptional activation of the IL-8 gene by NF-kappa B p65 (RelA) and NF-IL-6. J Immunol 1994;153(1):153–64. [PubMed] [Google Scholar]
- 16.Azimi N, Brown K, Bamford RN, Tagaya Y, Siebenlist U, Waldmann TA. Human T cell lymphotropic virus type I Tax protein trans-activates interleukin 15 gene transcription through an NF-kappaB site. Proc Natl Acad Sci U S A 1998;95(5):2452–7 doi 10.1073/pnas.95.5.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiscott J, Marois J, Garoufalis J, D'Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol 1993;13(10):6231–40 doi 10.1128/mcb.13.10.6231-6240.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem 2005;95(3):497–505 doi 10.1002/jcb.20477. [DOI] [PubMed] [Google Scholar]
- 19.Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville MP, et al. NF- kappa B2/p100 induces Bcl-2 expression. Leukemia 2003;17(7):1349–56 doi 10.1038/sj.leu.2402982. [DOI] [PubMed] [Google Scholar]
- 20.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol 2000;20(8):2687–95 doi 10.1128/MCB.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res 2008;68(16):6762–9 doi 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bono JS, Guo C, Gurel B, De Marzo AM, Sfanos KS, Mani RS, et al. Prostate carcinogenesis: inflammatory storms. Nat Rev Cancer 2020;20(8):455–69 doi 10.1038/s41568-020-0267-9. [DOI] [PubMed] [Google Scholar]
- 23.Liu B, Li L, Yang G, Geng C, Luo Y, Wu W, et al. PARP Inhibition Suppresses GR-MYCN-CDK5-RB1-E2F1 Signaling and Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer. Clin Cancer Res 2019;25(22):6839–51 doi 10.1158/1078-0432.CCR-19-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu C, Peng S, Pilie PG, Geng C, Park S, Manyam GC, et al. PARP and CDK4/6 Inhibitor Combination Therapy Induces Apoptosis and Suppresses Neuroendocrine Differentiation in Prostate Cancer. Mol Cancer Ther 2021;20(9):1680–91 doi 10.1158/1535-7163.MCT-20-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39(4):782–95 doi 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 2006;177(10):7303–11 doi 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553(7686):91–5 doi 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015;347(6227):aaa2630 doi 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 29.Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep 2017;20(8):1818–29 doi 10.1016/j.celrep.2017.07.075. [DOI] [PubMed] [Google Scholar]
- 30.Manore SG, Doheny DL, Wong GL, Lo HW. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front Oncol 2022;12:866014 doi 10.3389/fonc.2022.866014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee GT, Kwon SJ, Lee JH, Jeon SS, Jang KT, Choi HY, et al. Macrophages induce neuroendocrine differentiation of prostate cancer cells via BMP6-IL6 Loop. Prostate 2011;71(14):1525–37 doi 10.1002/pros.21369. [DOI] [PubMed] [Google Scholar]
- 32.Hsia HC, Hutti JE, Baldwin AS. Cytosolic DNA Promotes Signal Transducer and Activator of Transcription 3 (STAT3) Phosphorylation by TANK-binding Kinase 1 (TBK1) to Restrain STAT3 Activity. J Biol Chem 2017;292(13):5405–17 doi 10.1074/jbc.M116.771964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018;15(4):234–48 doi 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.U'Ren L, Guth A, Kamstock D, Dow S. Type I interferons inhibit the generation of tumor-associated macrophages. Cancer Immunol Immunother 2010;59(4):587–98 doi 10.1007/s00262-009-0776-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, et al. Androgen receptor inhibitor-induced "BRCAness" and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal 2017;10(480) doi 10.1126/scisignal.aam7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, et al. Molecular cloning of a ligand for the inducible T cell gene 4–1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. Eur J Immunol 1993;23(10):2631–41 doi 10.1002/eji.1830231037. [DOI] [PubMed] [Google Scholar]
- 37.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun 2011;12(6):399–414 doi 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S, Chu C, Wu Z, Liu F, Xie J, Yang Y, et al. IFIH1 Contributes to M1 Macrophage Polarization in ARDS. Front Immunol 2020;11:580838 doi 10.3389/fimmu.2020.580838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braune J, Weyer U, Hobusch C, Mauer J, Bruning JC, Bechmann I, et al. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J Immunol 2017;198(7):2927–34 doi 10.4049/jimmunol.1600476. [DOI] [PubMed] [Google Scholar]
- 40.McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, et al. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A 2005;102(27):9589–94 doi 10.1073/pnas.0501794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 2016;14(2):282–97 doi 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep 2017;7:39858 doi 10.1038/srep39858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Pleasure D, Deng W, Guo F. Therapeutic Potentials of Poly (ADP-Ribose) Polymerase 1 (PARP1) Inhibition in Multiple Sclerosis and Animal Models: Concept Revisiting. Adv Sci (Weinh) 2022;9(5):e2102853 doi 10.1002/advs.202102853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang F, Zhao M, Chang B, Zhou Y, Wu X, Ma M, et al. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol Cell 2022;82(11):2032–49 e7 doi 10.1016/j.molcel.2022.03.034. [DOI] [PubMed] [Google Scholar]
- 45.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14(7):399–416 doi 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kfoury Y, Baryawno N, Severe N, Mei S, Gustafsson K, Hirz T, et al. Human prostate cancer bone metastases have an actionable immunosuppressive microenvironment. Cancer Cell 2021;39(11):1464–78 e8 doi 10.1016/j.ccell.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chow A, Schad S, Green MD, Hellmann MD, Allaj V, Ceglia N, et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell 2021;39(7):973–88 e9 doi 10.1016/j.ccell.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zarif JC, Baena-Del Valle JA, Hicks JL, Heaphy CM, Vidal I, Luo J, et al. Mannose Receptor-positive Macrophage Infiltration Correlates with Prostate Cancer Onset and Metastatic Castration-resistant Disease. Eur Urol Oncol 2019;2(4):429–36 doi 10.1016/j.euo.2018.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun 2019;10(1):100 doi 10.1038/s41467-018-07927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gruber JJ, Chen J, Geller B, Jager N, Lipchik AM, Wang G, et al. Chromatin Remodeling in Response to BRCA2-Crisis. Cell Rep 2019;28(8):2182–93 e6 doi 10.1016/j.celrep.2019.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data for RM-1-BM cell lines and RAW264.7 cells used in coculture model experiments were deposited to The Gene Expression Omnibus (GSE240199).