

Abstract
Replacement of the lost myelin sheath is a therapeutic goal for treating demyelinating diseases of the central nervous system (CNS), such as multiple sclerosis (MS). The G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor (GPCR) GPR17, which is phylogenetically closely related to receptors of the “purinergic cluster,” has emerged as a modulator of CNS myelination. However, whether GPR17-mediated signaling positively or negatively regulates this critical process is unresolved. We identified a small-molecule agonist, MDL29,951, that selectively activated GPR17 even in a complex environment of endogenous purinergic receptors in primary oligodendrocytes. MDL29,951-stimulated GPR17 engaged the entire set of intracellular adaptor proteins for GPCRs: G proteins of the Gαi, Gαs, and Gαq subfamily, as well as β-arrestins. This was visualized as alterations in the concentrations of cyclic adenosine monophosphate and inositol phosphate, increased Ca2+ flux, phosphorylation of extracellular signal–regulated kinases 1 and 2 (ERK1/2), as well as multifeatured cell activation recorded with label-free dynamic mass redistribution and impedance biosensors. MDL29,951 inhibited the maturation of primary oligodendrocytes from heterozygous but not GPR17 knockout mice in culture, as well as in cerebellar slices from 4-day-old wild-type mice. Because GPCRs are attractive targets for therapeutic intervention, inhibiting GPR17 emerges as therapeutic strategy to relieve the oligodendrocyte maturation block and promote myelin repair in MS.
INTRODUCTION
Multiple sclerosis (MS) is a severe neurological disease characterized by autoimmune-mediated demyelination, oligodendrocyte damage, and, ultimately, axonal loss (1-5). Demyelination initially impairs rapid saltatory nerve conduction and can cause axonal degeneration followed by progressive and irreversible functional deficits and neurological disability if not repaired through remyelination, a complex process that forms new myelin sheaths along axon tracts (1-5). Despite an increasing appreciation of the importance of remyelination, most current medicines for MS are immunomodulatory drugs targeted against the inflammatory component of the disease (4, 6-8). Furthermore, the complex regulatory mechanisms underlying the remyelination process are poorly understood, and it is not clear why remyelination is inadequate or absent in MS (2-4, 9). Oligodendrocyte precursor cells (OPCs) are present in demyelinating lesions and normally foster the repair process. They do so by opposing the action of intrinsic oligodendrocyte differentiation inhibitors (ID proteins), such as ID2 or ID4, thereby allowing OPCs to progress toward mature myelin-forming oligodendrocytes (2-4). Promoting myelin repair is emerging as a therapeutic strategy but is not yet exploited therapeutically, which may be because of—at least in part—the difficulties in targeting oligodendrocyte differentiation inhibitors with small-molecule drugs (2, 3, 8). The only agent with the prospect of enhancing remyelination at present is a monoclonal antibody against LINGO-1 [leucine-rich repeat and immunoglobulin (Ig) domain-containing Nogo receptor interacting protein 1], a negative regulator of oligodendrocyte differentiation and myelination (10, 11).
A class of membrane proteins with great success as targets for small-molecule ligand discovery is the family A G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptors (GPCRs) (12, 13). GPR17 is an orphan family A GPCR that is phylogenetically related to purinergic and cysteinyl-leukotriene (CysLT) receptors (14). It was identified by a transcriptomic approach using central nervous system (CNS) samples from myelination-deficient mice to be a cell-intrinsic timer that controls the transition of oligodendrocytes from the immature to the mature myelinating stage (15). GPR17 is abundant in differentiating OPCs in a temporally controlled manner (15-17). Mice overexpressing GPR17 in oligodendrocytes display characteristic features of demyelinating diseases, whereas mice genetically lacking GPR17 show premature myelination (15). Contrary to these findings from genetic studies, small interfering RNA (siRNA)–based gene silencing experiments and pharmacological studies applying the purported endogenous agonists for this receptor, uracil nucleotides and CysLTs, support the notion that GPR17 activation promotes oligodendrocyte differentiation and progression toward mature myelin-forming cells (16-18). Hence, there is therapeutic promise for GPR17 modulators to treat pathologies associated with myelin repair in CNS demyelinating diseases, but it is unresolved whether activation or inhibition is the desired therapeutic principle. Both endogenous ligand classes are unsuited to differentiate between the functions of purinergic receptors, CysLT receptors, and GPR17 ex vivo or in vivo where multiple receptors often coincide (18, 19). Moreover, several independent reports do not support the notion that GPR17 represents the elusive dualistic orphan receptor (20-24). Consequently, using bioinformatics and biomolecular screening assays in cells and in vivo validation, we set out to search for compounds that reliably and specifically activate GPR17 to solve the current controversy on the role of this receptor in oligodendrocyte myelination.
RESULTS
A small-molecule agonist of the orphan GPR17 unveils pleiotropic signaling in heterologous expression systems
GPR17 is phylogenetically related to both purinergic (P2Y) nucleotide and CysLT receptors (14), but it also has critical, positively charged residues—the characteristic H-X-X-R/K motif in the transmembrane domain VI—in common with receptors for small carboxylic acids (fig. S1). We therefore considered the possibility that GPR17 might respond to an endogenous modulator with at least one negatively charged moiety and established a compound repository consisting of precursors and intermediates of metabolic pathways and structurally related ligands with known and unknown biological activities, but with a particular focus on neuroactive compounds (table S1). These compounds, including the purported endogenous activators (14), were characterized for agonist activity in human embryonic kidney (HEK) 293 cells stably transfected with a hemagglutinin (HA) epitope–tagged version of the short isoform of human GPR17, hereafter referred to as GPR17. This isoform was chosen because it is the only ortholog expressed in rodents, which serve as a source for cells and tissues to explore the physiological function of GPR17. Before functional analysis of the selected ligands, the abundance and trafficking to the cell surface of GPR17 in the transfected HEK cells were confirmed by ELISA (enzyme-linked immunosorbent assay) and immunocytochemical staining (fig. S2).
To increase the likelihood of identifying GPR17 activators, we took advantage of a pathway-unbiased, yet pathway-sensitive, label-free assay technology, which detects dynamic mass redistribution (DMR) that occurs as a consequence of cell stimulation irrespective of the particular GPCR signaling pathway involved (25-27). Using this noninvasive methodology, we identified two compounds—MDL29,951 and GV150526A (Fig. 1A)—that resulted in robust DMR traces in cells expressing GPR17 (Fig. 1, B to D). Native HEK cells were unresponsive to MDL29,951 (Fig. 1, D and E) but were sensitive to stimulation with carbachol and ATP (adenosine 5′-triphosphate), which served as viability controls through activation of endogenous muscarinic and P2Y receptors, respectively (fig. S3). Of particular interest was MDL29,951 because of its higher potency and the complete lack of effect in native HEK cells as opposed to GV150526A, which bypasses the strict requirement of GPR17 for cell activation (Fig. 1D). Furthermore, only MDL29,951, but not GV150526A or any of the putative endogenous agonists, robustly stimulated the long isoform of GPR17 (Fig. 1E and fig. S4). For this reason, MDL29,951 was prepared by chemical synthesis to explore GPR17 behavior in more detail.
Fig. 1. Structures and functional properties of GPR17 agonists MDL29,951 and GV150526Ain HEK-hGPR17 cells.
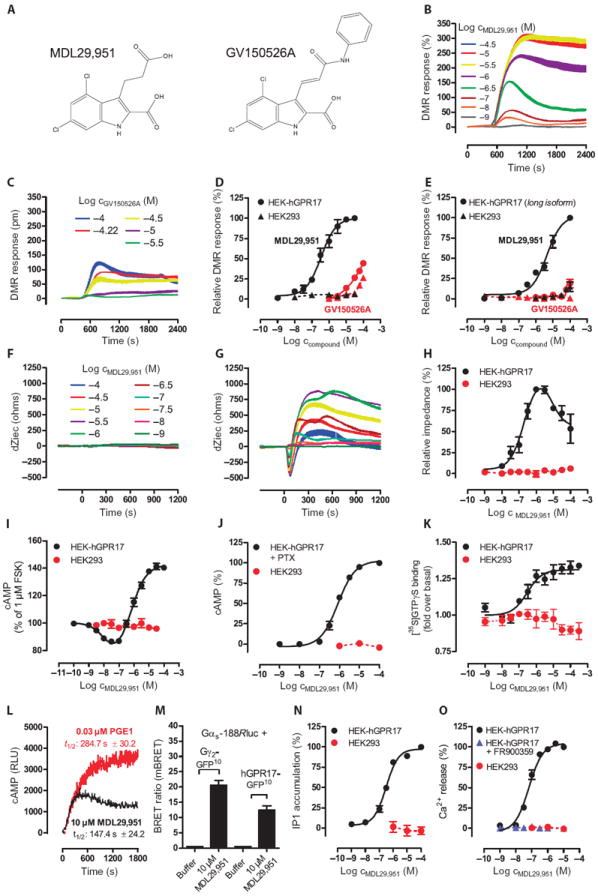
(A) Chemical structures of GPR17 agonists. (B and C) Representative traces of MDL29,951 (B) andGV150526A (C) determined by label-free DMR assays in HEK cells overexpressing GPR17. (D) Concentration-effect curves derived from the traces in (B) and (C). (E) Functional activity of MDL29,951 of the long isoform of hGPR17 in DMR assays in transfected or untransfected HEK cells. (F and G) MDL29,951-mediated whole-cell responses in native HEK293 (F) and HEK-hGPR17 cells (G) assessed by label-free bioimpedance sensing. Data are representative traces depicted as changes of extracellular current (dZiec). (H) Concentration-effect relationships from the traces in (F) and (G). (I to O) MDL29,951-mediated GPR17 activation in assays recording (I) the inhibition of forskolin-stimulated adenylyl cyclase, (J) cAMP elevation in the presence of PTX, (K) [35S]GTPγS binding to the indicated membranes, (L) real-time monitoring of cAMP accumulation, (M) direct interaction between the indicated BRET2 partners, (N) intracellular inositol phosphate (IP1) accumulation, and (O) Ca2+ flux in the presence and absence of the Gαq inhibitor FR900359. (B, C, F, G, and L) Data are representative of at least n = 3 independent experiments. (D, E, H to K, N, and O) Data are means ± SEM, n = 3 to 6. (M) Data are means + SEM, n = 5 to 6.
To corroborate the findings from our DMR studies, we used a second label-free technology platform called CellKey that translates pharmacologically mediated alterations of cellular activity into bioimpedance signals (28). CellKey recordings revealed concentration-dependent and multifeatured whole-cell responses to MDL29,951, characterized by an initial transient negative phase followed by a rapid ascending phase that reached a maximum, after which impedance slowly declined. This complex, time-resolved pattern of impedance recordings most likely reflected real-time integration of multiple signaling events that strictly required the presence of GPR17 (Fig. 1, F to H).
Because label-free recordings are multidimensional, we wondered which of the individual GPCR-associated signaling cascades are accessible to GPR17. To this end, we monitored GPR17-mediated cell activation using assays that focus on single functional endpoints. cAMP (adenosine 3′, 5′-monophosphate) accumulation assays illustrate a bell-shaped concentration-effect curve in response to MDL29,951, suggesting dual modulation of adenylyl cyclase that is either inhibited or stimulated by Gαi and Gαs proteins, respectively (Fig. 1I). In agreement with this notion, pretreatment of cells with the Gαi inhibitor pertussis toxin (PTX) abolished MDL29,951-mediated attenuation of cAMP formation and isolated the cAMP-increasing capacity of GPR17 (Fig. 1J). Engagement of Gαi proteins by GPR17 was further corroborated in assays that quantify incorporation of [35S]GTPγS into the α subunit of heterotrimeric G proteins (Fig. 1K). Increased cAMP concentrations at higher agonist concentrations reflected true GPR17-Gαs interaction rather than activation of an unknown Gαs-sensitive receptor by a secreted ligand, because (i) cAMP accumulation occurred over a time course comparable to that of prostaglandin E1, a bona fide stimulus of a Gαs-sensitive receptor (29) (Fig. 1L), and (ii) a bioluminescence resonance energy transfer (BRET2) assay demonstrated that both direct ligand-induced interactions between GPR17 and Gαs as well as rapid molecular rearrangement within the Gαs-βγ heterotrimer occurred after MDL29,951 treatment (Fig. 1M). Relevance of functional Gαq activity was evident from inositol phosphate accumulation (Fig. 1N) and intracellular Ca2+ mobilization because GPR17-mediated Ca2+ flux was blunted by the Gαq-selective inhibitor FR900359 (30, 31) (Fig. 1O). Productive Gαq coupling of MDL29,951-stimulated GPR17 led us to examine whether a Ca2+-promoted mechanism may cooperate with Gαs proteins to achieve maximal stimulation of adenylyl cyclase. Indeed, accumulation of cAMP is attenuated when Gαq proteins are inhibited, both in the presence (Gαi and Gαs signaling visible; fig. S5A) and absence (only Gαs signaling visible; fig. S5B) of forskolin. Together, our results indicate that MDL29,951-stimulated GPR17 activates the entire repertoire of pathways linked to second messenger production and initiates a rather pleiotropic biological response. No alteration of second messengers was observed when native HEK cells were exposed to MDL29,951, confirming that it required GPR17 to induce a functional effect (Fig. 1, I, J, N, and O). In contrast to the ability of MDL29,951 to activate GPR17, a set of five small-molecule GPR17 agonists (ASINEX ligands 1 to 5), identified in a virtual screening approach (32), was inactive in both label-free DMR and classical Ca2+ mobilization assays (fig. S6).
To ascertain the validity of our GPR17 agonist, we also recorded integrated live cell responses and the activation of defined second messenger cascades in two additional cell lines that were engineered to stably express GPR17: Chinese hamster ovary (CHO)–hGPR17 and human astrocytoma 1321N1-hGPR17 cells. Both lines responded with robust and multifaceted whole-cell responses in DMR assays (Fig. 2, A to D) or bioimpedance (Fig. 2, E to H), respectively, after treatment with MDL29,951. Akin to the results obtained in HEK-hGPR17 cells, the incorporation of [35S]GTPγS into Gα proteins reflected Gαi coupling, and the bell-shaped concentration-effect curve demonstrated dual modulation of cAMP accumulation, in which low agonist concentrations attenuated and high concentrations increased formation of cAMP, in both CHO-hGPR17 cells (Fig. 2, I and J) and 1321N1-hGPR17 cells (Fig. 2, K and L). Pretreatment of CHO-hGPR17 cells with the Gαi inhibitor PTX abolished the Gαi component of the concentration-effect curve (Fig. 2M). In these cells, production of cAMP was slightly attenuated by pharmacological inhibition of Gαq proteins (fig. S7A), despite robust engagement of the Gαq signaling cascade (Fig. 2N). In contrast, 1321N1-hGPR17 cells did not retain the capacity to produce cAMP in the presence of PTX (Fig. 2O), indicating that cAMP synthesis requires prior stimulation of Gαi proteins in this cellular background. Furthermore, production of cAMP was moderately attenuated when Gαq proteins were pharmacologically inhibited (fig. S7B), despite also showing robust induction of the Gαq signaling cascade (Fig. 2P). These data illustrate that the increase in cAMP relies on a functional interaction between GPR17 and Gαs proteins with minimal (CHO-hGPR17) or moderate (1321N1-hGPR17) contribution from a Ca2+-dependent mechanism. A comprehensive compilation of MDL29,951 potencies across all three GPR17 cell lines is given in table S2. Together, our results demonstrate that (i) GPR17 can be functionally and stably expressed in various cellular backgrounds, (ii) MDL29,951-stimulated GPR17 signals through heterotrimeric G proteins from different families in a cell context–dependent manner, and (iii) MDL29,951, but not ASINEX ligands 1 to 5, reliably and reproducibly activates this orphan receptor.
Fig. 2. MDL29,951 robustly activates cellular signaling in CHO-hGPR17 and 1321N1-hGPR17 cells.
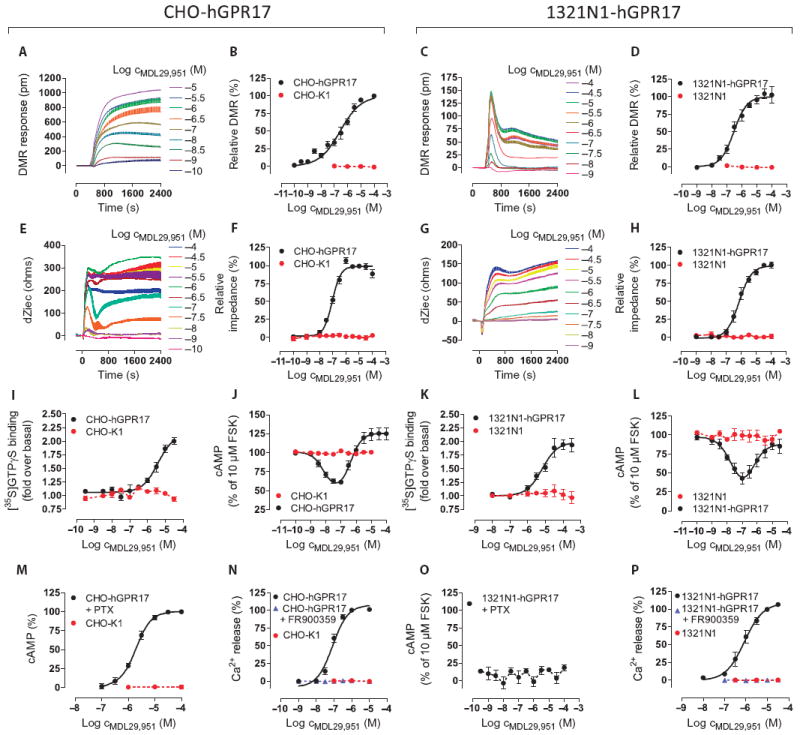
(A to H) Label-free DMR (A to D) and label-free bioimpedance (quantified as alteration of extracellular current dZiec) (E to H) in CHO-hGPR17 and 1321N1-hGPR17 cells treated with the indicated concentrations of MDL29,951. Data are representative traces from at least three independent experiments, or means ± SEM from (B and D) 3 to 10 independent experiments or (F and H) 4 to 5 independent experiments, each performed in triplicate. (I to L) Incorporation of [35S]GTPγS binding into Gα subunits and inhibition of forskolin (FSK)–stimulated cAMP production in CHO-hGPR17 cells (I and J, respectively) or 1321N1-hGPR17 cells (K and L, respectively) treated with MDL29,951. (M to P) As a measure of GPR17 functionality, accumulation of cAMP and mobilization of Ca2+ from intracellular stores in CHO-hGPR17 cells (M and N, respectively) or 1321N1-hGPR17 cells (O and P, respectively) treated withMDL29,951 were quantified. (I to P) Data are means ± SEM from three to seven independent experiments, each performed in triplicate.
GPR17 recruits β-arrestin2 in an agonist-dependent manner
GPCR signaling events of interest are those set into motion by heterotrimeric G proteins or arrestins (33-41). Unlike heterotrimeric G proteins, which are involved in second messenger production, arrestins may mediate G protein–independent signaling by acting as scaffolds for signaling cascade components such as members of the mitogen-activated protein kinase (MAPK) family (33-41). Functional characterization of GPR17 indicated that multiple G protein pathways are engaged, but whether G protein activation is followed by β-arrestin recruitment or whether β-arrestin recruitment occurs in its own right has not yet been reported. Detection of receptor-arrestin interaction was measured by BRET2 assays by fusing the energy donor Renilla reniformis luciferase enzyme (Rluc) to GPR17 and the energy acceptor green fluorescent protein 2 (GFP2) to β-arrestin2. The GPR17-Rluc fusion protein properly trafficked to the plasma membrane (fig. S8, A and B) and retained functional activity as evidenced in holistic DMR (fig. S8, C and D) and classical second messenger assays (fig. S8, E and F). The kinetics of GPR17-arrestin recruitment after treatment with agonist was quantified over time. Real-time monitoring of agonist-induced BRET2 showed immediate arrestin recruitment upon agonist addition (Fig. 3A), with a maximum recruitment occurring at about 1 min and then gradually declining but still detectable even after 60 min in the presence of MDL29,951 (Fig. 3B). These data support the notion that the receptor and arrestin interact in a sustained manner. Agonist-promoted arrestin recruitment quantified 5 min after agonist treatment was concentration-dependent with a potency similar to that determined in the G protein activation assays (Fig. 3C and fig. S8, D to F). This may suggest that arrestin recruitment occurs as a consequence of G protein signaling. Yet, under conditions that completely interdicted G protein activation of GPR17 (Fig. 3, D to F), MDL29,951-induced recruitment of arrestin to the receptor was still detectable, albeit at decreased amounts (Fig. 3G). Therefore, MDL29,951-stimulated GPR17 generates typical G protein responses and recruits arrestin through dual mechanisms, dependently or independently of G protein activity.
Fig. 3. MDL29,951 stimulates hGPR17 in β-arrestin2 recruitment, phospho-ERK1/2, and internalization assays.
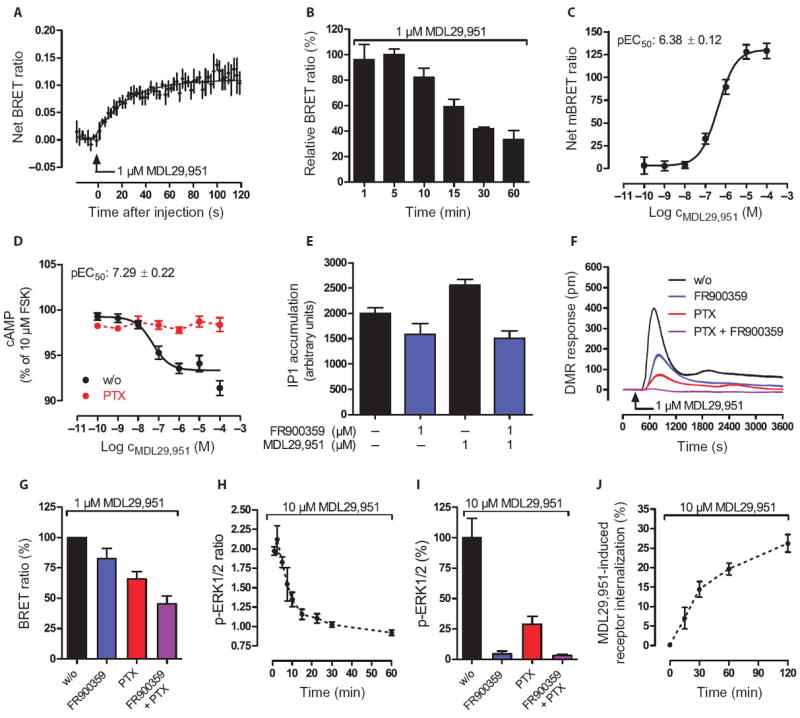
(A to C) BRET2 assays between hGPR17-Rluc and GFP2–β-arrestin2 in stably transfected HEK293 cells measured (A) as real-time kinetics, (B) at time intervals, or (C) at 5 min after the indicated MDL29,951 treatment. (D) Effect of PTX (50 ng/ml) on forskolin-stimulated cAMP production in HEK-BRET-hGPR17 cells. (E) Effect of the Gαq-selective inhibitor FR900359 on MDL29,951-induced inositol phosphate (IP1) production in HEK-BRET-hGPR17 cells. (F) DMR of Gαq and Gαi signaling in HEK-BRET-hGPR17 cells after pretreatment with FR900359 (1 μM) and PTX (50 ng/ml). (G) MDL29,951-stimulated BRET2 between hGPR17-Rluc and GFP2–β-arrestin2 in the absence (w/o) or presence of FR900359 (1 μM), PTX (50 ng/ml), or both. (H) Kinetics of the phosphorylation of ERK1/2 in HEK-BRET-hGPR17 cells after treatment with MDL29,951. (I) Phosphorylation of ERK1/2 in HEK-BRET-hGPR17 cells after MDL29,951 after pretreatment with FR900359 (1 μM), PTX (50 ng/ml), or both. (J) ELISA-based assessment of GPR17 internalization kinetics in HEK-hGPR17 cells treated with MDL29,951. (A and F) Representative BRET and DMR kinetics (means ± SE from three triplicates, representative of three independent experiments); all other data are means ±/+ SEM from two to six independent experiments.
GPR17 activation is followed by the phosphorylation of ERK and its internalization
G protein–independent recruitment of arrestin may serve to initiate a new wave of downstream signaling such as regulation of phosphorylation of members of the MAPK family, in particular extracellular signal–regulated kinases 1 and 2 (ERK1/2) (33, 34, 36-38, 42, 43). It has been suggested that G protein–dependent activation of MAPK is maximal at short time points (within 2 to 5 min), whereas β-arrestin–mediated activation is apparent at later time points (33). To examine whether activation of GPR17 precedes the phosphorylation of ERK1/2 and to investigate the underlying molecular mechanism, we used the same cellular system as applied before to examine GPR17-arrestin interaction. We detected robust ERK phosphorylation that was rapid, transient in nature (peaking within 2 min; Fig. 3H), and dependent on G protein signaling (Fig. 3I). Notably, ERK activation was eliminated by pretreatment of cells with the Gαq-selective inhibitor FR900359, yet ERK retained partial capacity to be phosphorylated in the presence of the Gαi inhibitor PTX. These results reveal an interesting hierarchy used by GPR17 to control cellular ERK1/2 activation: Gαq-mediated ERK activation occurs upstream to that mediated by Gαi. Together, our results provide evidence that MDL29,951-stimulated GPR17—despite its ability to recruit arrestin in a G protein–independent manner—heavily relies on G proteins to rapidly induce the phosphorylation of ERK1/2. Therefore, arrestin recruitment may mainly serve to desensitize the receptor and regulate its functional responsiveness or to facilitate receptor removal from the plasma membrane. Indeed, MDL29,951 appeared to limit the plasma membrane abundance of GPR17 because cells exposed to the agonist displayed pronounced loss of GPR17 surface expression (Fig. 3J).
GPR17 signaling can be silenced with inhibitors of the cysteinyl-leukotriene 1 receptor
The leukotriene receptor antagonists pranlukast and montelukast can inhibit GPR17 in the nanomolar concentration range (14, 44). Because we were unable to activate GPR17 with any of the published agonists, including the small-molecule ASINEX ligands (fig. S6), we were curious about the functional properties of the antagonists and subjected them to multiple cellular assays. Pranlukast, but not montelukast, inhibited GPR17 in a concentration-dependent manner, yet micromolar concentrations of pranlukast were required to completely abrogate GPR17 signaling in label-free DMR assays (Fig. 4A). This pattern of antagonist behavior was similar to that obtained in assays measuring inositol phosphate accumulation (Fig. 4B), but it deviated when mobilization of intracellular Ca2+—a very rapid nonequilibrium assay—was used as an activity reporter (Fig. 4C). Although discrepancies of antagonist pharmacology determined under equilibrium versus nonequilibrium conditions are well known (45), our data indicate that pranlukast is superior over montelukast in achieving functional inhibition of GPR17. Both antagonists effectively inhibited cysteinyl-leukotriene 1 (CysLT1) receptor signaling with potencies that agree with published affinities for these ligands (46) (fig. S9). The molecular nature of the pranlukast-hGPR17 interaction was further investigated by generating concentration-effect curves of inositol phosphate accumulation induced by MDL29,951 in the absence or presence of pranlukast (Fig. 4D). Increasing concentrations of pranlukast induced parallel rightward shifts in the effect of MDL29,951 without depressing the maximal response. This mode of surmountable antagonism is compatible with competitive interaction at a common binding site, yet the slope of the Schild regression was significantly steeper than unity (Fig. 4E), implying that additional molecular mechanisms are involved.
Fig. 4. CysLT1 receptor antagonists differentially inhibit human and rodent GPR17 orthologs in recombinant human HEK293 cells.
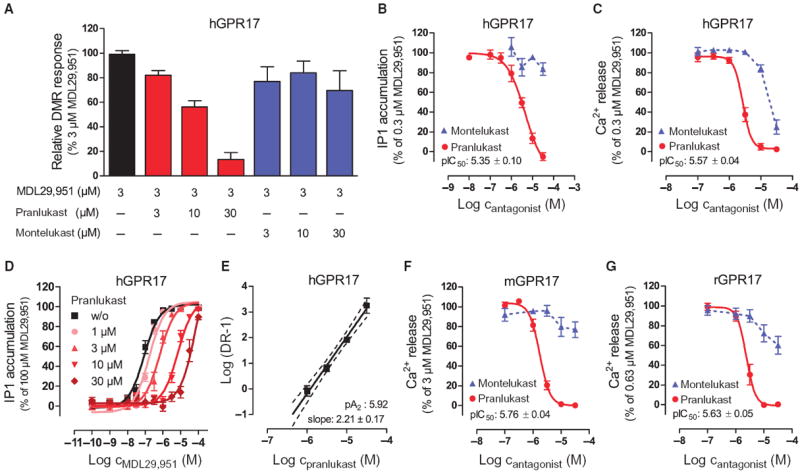
(A) Inhibition of MDL29,951-mediated hGPR17 activation by pranlukast and montelukast in label-free DMR assays. Data are means + SEM from 4 to 12 independent experiments, each performed in triplicate. (B and C) Concentration-effect profile of MDL29,951-stimulated hGPR17 cells of the indicated CysLT1 antagonists assessed by (B) IP1 or (C) Ca2+ mobilization. (D) MDL29,951 concentration-effect curves in the absence or presence of fixed concentrations of pranlukast assessed by IP1 accumulation. (E) Schild regression of the curves depicted in (D). (F and G) Effect of CysLT1 antagonists pranlukast and montelukast on MDL29,951-stimulated mGPR17 (F) and rGPR17 (G) Ca2+ mobilization. (B to G) Data are means ± SEM from three to six independent experiments, each performed in triplicate. (B, C, F, and G) MDL29,951 was used at concentrations that yielded about 80% of its maximal response in the respective assay and cell line.
MDL29,951 activates mouse and rat GPR17
Because marked differences of ligand potency exist among species-specific orthologs of GPCRs (47, 48), which precludes a simple translation of ligand pharmacology observed at the human receptor to ex vivo or in vivo animal models, we next examined whether MDL29,951 would be of value to elucidate the biological function of rodent GPR17 orthologs. Rat (r) and mouse (m) GPR17 were cloned and stably transfected into human HEK293 cells, and their proper plasma membrane localization was verified by immunocytochemical staining (fig. S10). Robust and concentration-dependent activation of rGPR17 and mGPR17 was observed in label-free DMR and classical endpoint assays upon application of MDL29,951 reminiscent of the signaling profile we obtained for human GPR17 in this cellular background (fig. S11). In agreement with our data on the human ortholog, no signs of activation were obtained when cells were challenged with ASINEX ligands (fig. S12), and only pranlukast, but not montelukast, effectively attenuated the function of MDL29,951-stimulated mouse and rat GPR17 (Fig. 4, F and G). Both antagonists were ineffective when applied alone (fig. S13) and did not diminish Ca2+ mobilization triggered by the endogenously expressed muscarinic M3 receptor (fig. S14), confirming the specific nature of GPR17 inhibition. Together, our data reveal that GPR17 agonist MDL29,951 and GPR17 antagonist pranlukast—despite its clear preference for the CysLT1 receptor—may be applied in vitro, in cells, or in vivo to interrogate the physiological function of GPR17 and its implication in human and rodent physiology.
MDL29,951 does not activate receptors of the “purinergic cluster”
One of the goals of identifying ligands for orphan receptors is to elucidate the role of receptor signaling in normal and disease physiologies. This often requires application of the novel ligands to primary cells where the receptor is expressed in its natural environment. For this reason, it is of utmost importance to define the selectivity profile of MDL29,951 before investigating it in a primary cell context. We therefore tested the functional capability of MDL29,951 to activate phylogenetically related P2Y and CysLT receptors (Fig. 5, A to I, and table S3) and selected phylogenetically distant biogenic amine (histamine and dopamine) receptors (fig. S15) using both label-free real-time DMR and classical endpoint assays, respectively. MDL29,951 showed strong selectivity for GPR17 over the other 11 GPCRs tested. A favorable selectivity profile of this ligand is further supported by the notion that no activity of MDL29,951 was detected in DMR-based “receptor panning” across 16 different cell lines (fig. S16), many of which are coincidentally abundant for multiple purinergic receptors.
Fig. 5. MDL29,951 does not productively interact with receptors of the purinergic cluster.
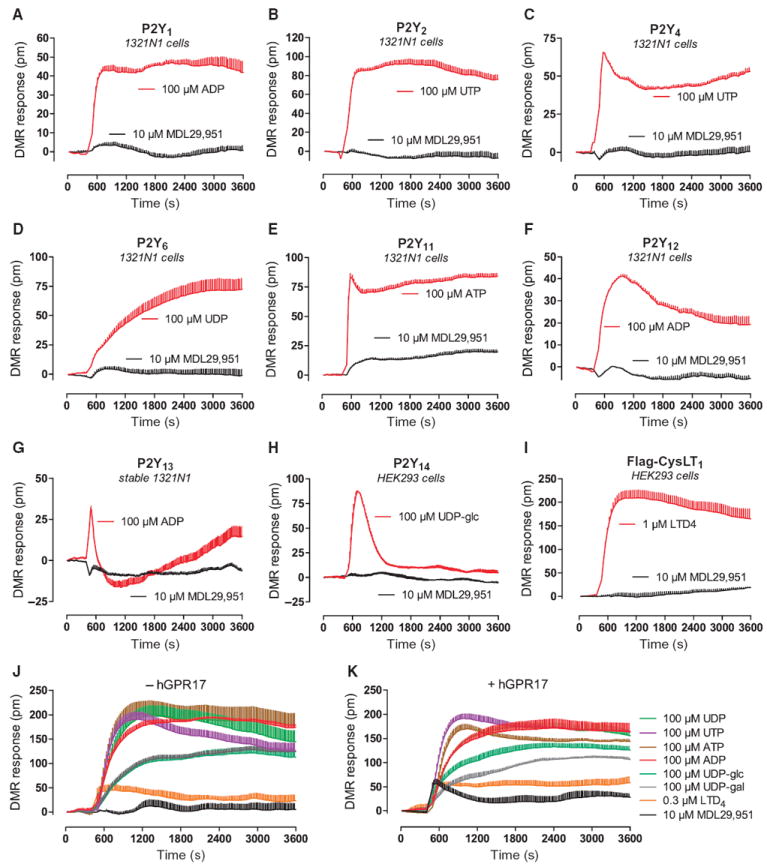
(A to I) Ability of MDL29,951 to trigger activation of the indicated receptors using whole-cell DMR assays as reporter for functional signaling capacity. Testing was performed along with an appropriate control stimulus for each cell line. Data in (A) to (I) are representative traces + SE of at least three independent experiments, each performed in triplicate. (J and K) Time course of whole-cell DMR recorded in HEK293 cells transiently transfected with a cocktail of P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14, and CysLT1 in the absence (J) or presence (K) of hGPR17 and treated with the ligands indicated in the legend to the side of (K). Data are traces + SE of technical triplicates representative of two to four independent experiments.
Even when a cocktail of eight purinergic receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14) was forcibly overexpressed in HEK293 cells [which are a host for several endogenous P2Y receptors (49-51); fig. S17], MDL29,951 remained inactive (Fig. 5J), unless GPR17 was also overexpressed (Fig. 5K). Successful transfection of complementary DNAs (cDNAs) of the cocktail was verified by responses of cells to the cognate receptor agonists (fig. S18). These results again confirm that MDL29,951 is remarkably selective and suggest its suitability to target GPR17 even in cells that have multiple purinergic receptors in endogenous or experimental conditions.
MDL29,951 activates GPR17 in primary oligodendrocytes
To assess the potential clinical relevance of this GPR17 agonist, we determined whether MDL29,951 retained the capacity to activate GPR17 in an endogenous system. We chose to study primary rat oligodendrocytes because GPR17 is abundant in these cells (15-17, 52). Primary rat oligodendrocyte cultures were established and characterized by immunocytochemistry using several specific oligodendrocyte markers [proteoglycan NG2 for OPCs, O antigen 4 (O4) for pre- and differentiated oligodendrocytes, and myelin basic protein (MBP) for ramified, mature oligodendrocytes] in addition to GPR17 (fig. S19). Consistent with previous data (17, 52), expression of GPR17 in spontaneously differentiating oligodendrocytes was temporally controlled. Therefore, GPR17 functionality was assessed at peak receptor expression, which occurred at day 4 in culture (fig. S19). MDL29,951 rapidly mobilized intracellular Ca2+ in a concentration-dependent manner (Fig. 6, A and B), a response that is undetectable in primary astrocytes, which did not have a detectable abundance of GPR17 (Fig. 6, B and C). MDL29,951-induced Ca2+ flux was GPR17-specific because it was counteracted by treatment with pranlukast (Fig. 6D), which did not affect carbachol-induced Ca2+ mobilization through endogenous M3 receptors (fig. S20). Pretreatment of primary oligodendrocytes with FR900359, but not PTX, prevented MDL29,951-induced Ca2+ flux, which suggests that an exclusively Gαq-dependent mechanism accounted for the observed Ca2+ release (Fig. 6E). Neither PTX nor FR900359 affected the viability of primary oligodendrocytes because Ca2+ flux by a nonreceptor stimulus was virtually unaltered when cells were pretreated with these inhibitors (fig. S21). MDL29,951 also decreased the amount of cAMP in GPR17-high oligodendrocytes, which have a high abundance of GPR17, but not in OPCs or primary astrocytes, which have low or no abundance of GPR17 (Fig. 6, C and F), and this effect in oligodendrocytes was abolished by pretreatment with PTX (Fig. 6F) or with pranlukast (Fig. 6G). Accumulation of cAMP was not apparent at higher concentrations of the GPR17 agonist (Fig. 6F; compare with fig. S11, C and G). This variability may be explained by differential coupling of GPR17 in engineered versus native expression systems or the higher receptor abundance in transfected cells, because it is well known that the same agonist may have only inhibitory or both inhibitory and stimulatory effects on cAMP production depending on receptor density (53). The data suggest that MDL29,951 can activate endogenous GPR17 in primary oligodendrocytes and that activated GPR17 engages both Gαi and Gαq, but not Gαs signaling pathways.
Fig. 6. MDL29,951 specifically activates GPR17 in primary rat oligodendrocytes.
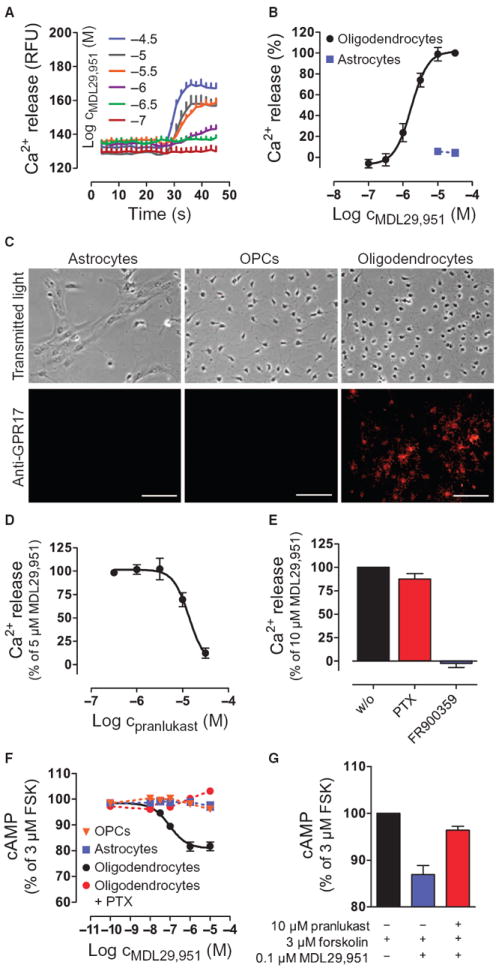
(A and B) Measure of the release of intracellular Ca2+ in oligodendrocytes and primary astrocytes treated with MDL29,951 over time (A) and concentration-effect relations derived from the traces in (A) for oligodendrocytes and for primary astrocytes (B). (C) Abundance of GPR17 in primary rat astrocytes, OPCs, and oligodendrocytes. Scale bars, 50 μm. (D) MDL29,951-mediated Ca2+ release in the presence of GPR17 antagonist pranlukast. (E) MDL29,951-stimulated Ca2+ release in the presence of the Gαi inhibitor PTX or the Gαq inhibitor FR900359. (F) Effect of MDL29,951 on forskolin-stimulated cAMP production in OPCs, astrocytes, or oligodendrocytes in the absence or presence of the Gαi inhibitor PTX. (G) MDL29,951-mediated cAMP inhibition in the absence and presence of the GPR17 antagonist pranlukast. (A and C) Data are representative of at least three independent experiments; (B and D to G) data are means ±/+ SEM of at least three independent experiments.
Activation of GPR17 signaling negatively regulates oligodendrocyte maturation
Gene silencing experiments using GPR17-specific siRNAs suggest that receptor inhibition impairs the oligodendroglia differentiation program (17). Genetic evidence from mice overexpressing or deficient in GPR17, on the other hand, supports the notion that inhibition of GPR17 is critical for oligodendrocyte terminal differentiation and myelination (15). Hence, it is unresolved whether GPR17 is a “promotor” or a “brake” for initiation of the remyelination program, an issue with great implication for development of remyelination therapeutics. Discovery of the first synthetic and selective GPR17 agonist that retains the capacity to activate the receptor in its natural environment provided an opportunity to resolve the enigmatic role of GPR17 in oligoden-drogliogenesis. Therefore, we treated oligodendrocytes from GPR17 heterozygous (GPR17+/−) and null (GPR17−/−) mice with MDL29,951 to clarify this discrepancy. Notably, GPR17+/− mice have similar abundance of GPR17 compared with wild-type littermates (15) but contain nuclear-localized GFP in the GPR17 locus to facilitate cell tracking in differentiation studies. In agreement with a role for GPR17 in inhibiting oligodendrocyte maturation (15), a significant increase in the number of mature, MBP-expressing oligodendrocytes was encountered in cultures from GPR17−/− mice (Fig. 7, A and B). In cultures from heterozygous (GPR17+/−), but not homozygous (GPR17−/−), mice, MDL29,951 markedly attenuated the capacity of oligodendrocytes to differentiate (Fig. 7, A, C, and D), evident from the reduced abundance of MBP, the decreased percentage of MBP-positive cells, and the absence of elaborated ramifications in most GPR17+/− cells (Fig. 7, A and C to E). Notably, because MDL29,951 did not alter the phenotype, number, or differentiation capacity of GPR17−/− oligodendrocytes (Fig. 7, A and D), the data support the notion that MDL29,951 does not induce off-target effects in this primary cell system. In accordance with the above observations, attenuation of GPR17 function with pranlukast had the opposite effect and enhanced the number of differentiated, mature oligodendrocytes in cultures from GPR17+/− but not knockout mice (Fig. 7, F to H). Arrest of the oligodendrocyte maturation program through the activation of GPR17 was also evident when cerebellar slice cultures from postnatal day 4 (P4) mice were exposed to MDL29,951, which resulted in pronounced loss of MBP-positive cells compared with control slices exposed to solvent (Fig. 7I). Together, these data provide evidence that both the presence and activation of GPR17 inhibit the oligodendrocyte maturation program and that MDL29,951 arrests oligodendrocytes at the less differentiated stage by specifically activating GPR17.
Fig. 7. Specific activation by MDL29,951 of GPR17 arrests oligodendrocytes at a precursor stage and impairs differentiation and maturation.
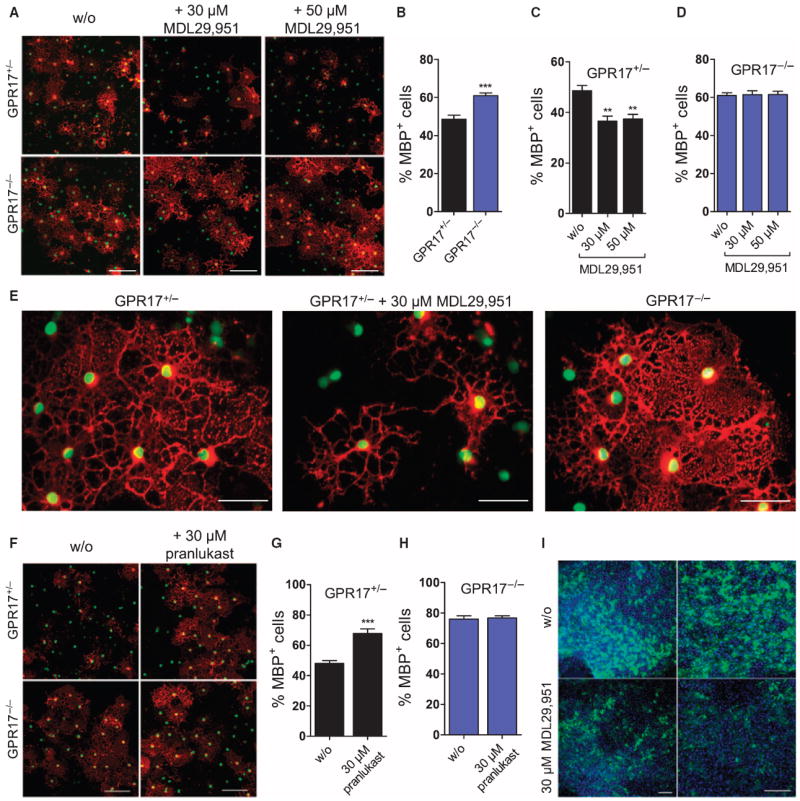
(A) OPCs isolated from GPR17+/− and GPR17−/− mice were cultured in the absence or presence of MDL29,951 for 3 days and stained for GFP to identify nuclei (green) and MBP to detect mature oligodendrocytes (red). (B to D) Quantification of the images shown in (A); data are means + SEM of MBP-positive OPCs obtained from OPCs cultured from at least three age-matched heterozygous and knockout littermates, each in two parallel cultures. (E) Higher-resolution image illustrating the effect of MDL29,951 on oligodendrocyte maturation in cultures from GPR17+/− and GPR17−/− mice. (F) Detection of GFP and MBP (red) in OPCs isolated from representative GPR17+/− and GPR17−/− mice and cultured in the absence and presence of 30 μM pranlukast for 4 days. (G and H) Quantifications of data in (F) are the means + SEM of MBP-positive OPCs obtained from at least three age-matched heterozygous or knockout littermates, each in two parallel cultures. (I) Cerebellar slice cultures from P4 wild-type mice were treated with/without MDL29,951 (30 μM) for 3 days, stained for MBP (green), and counterstained with DAPI (4′,6-diamidino-2-phenylindole). Scale bars, 100 μm (A, F, and I) or 50 μm (E). Statistical significance was analyzed by two-tailed, unpaired Student’s t test: **P < 0.01 and ***P < 0.001.
DISCUSSION
Demyelination, the loss of the multilayered membrane of insulation wrapped around axons produced and maintained by specialized oligodendrocytes, is a pathological process (1-5). In MS, demyelination of axons in the CNS is usually a consequence of autoimmune-mediated inflammatory injury to oligodendrocytes (1-4, 7). Remyelination is the process by which demyelinated axons are wrapped with new myelin sheaths produced by oligodendrocytes that have matured from OPCs in the vicinity of the demyelinating lesions. Thus, remyelination—the default “repair” response to a demyelinating lesion—can be viewed as a highly regenerative process coordinated and executed by oligodendrocytes, which fails or is inadequate in MS (1-5).
Oligodendrocyte differentiation and maturation are tightly regulated during development as well as during repair of demyelinating lesions, with multiple positive and negative factors ensuring spatiotemporal control of myelination at the appropriate locations (2-5, 9). Unraveling the molecular details that underlie myelination and remyelination after injury will certainly provide critical clues to understand how remyelination may be enhanced therapeutically. A particularly attractive prospect—froma therapeutic perspective—would be to block the function of endogenous inhibitors of myelin repair, yet no single therapeutic agent is available to date that achieves this goal.
GPR17 is an orphan GPCR that is abundant in the brain (23, 44, 54), particularly in the oligodendrocyte lineage cells of the CNS (15, 55). It has recently emerged as a key player orchestrating oligodendrocyte differentiation and maturation (15-18). Despite the strong evidence supporting a role for GPR17 in oligodendrocyte differentiation, it is unclear whether GPR17 promotes or inhibits transition of oligodendrocytes from the immature to the mature myelinating stage. The pharmacological tools available thus far, uracil nucleotides and CysLTs, are not suited to clarify this discrepancy because neither ligand class is competent to differentiate between the functions of purinergic receptors, CysLT receptors, and GPR17 ex vivo or in vivo where multiple receptors are often coincident, as is also the case in oligodendrocytes (17, 19). However, more importantly, there is no consensus on the validity of these ligands as GPR17 agonists, because the dualistic activation of GPR17 by the proposed endogenous ligands is difficult to recapitulate (20-22, 24, 56). The original deorphanizing report (14) and follow-up studies from the same group (16, 44, 57, 58) introduced the possibility that GPR17 accounts for the biological effects triggered by two classes of endogenous mediators: CysLTs and uracil nucleotides. Additional independent studies have probed the pharmacology of GPR17 in more depth, but despite this, GPR17 does not yet receive official recognition as being partnered with its true endogenous ligand (24, 56). Benned-Jensen and Rosenkilde (23) report a different agonist profile for nucleotide ligands but failed to recapitulate activation by CysLTs, a finding that is in line with other studies that reported lack of agonist activity altogether (20-22, 24) as well as our data in this study. Nevertheless, even a set of five small-molecule agonists identified by virtual screening and proposed to activate GPR17 with high potency (ASINEX ligands 1 to 5) (32) was inactive in our hands and has not been more widely used as research tool for this receptor. Clearly, the pharmacology and function of GPR17 remain rather contentious issues that need to be resolved with a ligand that reliably and reproducibly activates GPR17.
Guided by sequence alignments of receptors with similarity in the seven-transmembrane binding pocket, we compiled a repository of endogenous mediator compounds (precursors and intermediates of metabolic pathways) and structurally related ligands with a particular focus on those known to be involved in both neuroprotective and neurodegenerative processes. We identified the small-molecule MDL29,951, which activated the orphan GPR17 with notable selectivity and across species. Although it remains to be clarified whether agonism by MDL29,951 is orthosteric or allosteric in nature, and whether it stabilizes the same active states as does the elusive endogenous ligand, it does link GPR17 to all major effector pathways and therefore offers a new opportunity to probe signaling and function of this orphan receptor.
Herein, we took advantage of MDL29,951 to decode (i) the signaling of GPR17 and its rodent orthologs in recombinant and primary cells, and (ii) the enigmatic function of GPR17 in controlling oligodendrocyte maturation. The results of our study are entirely consistent with previous observations using GPR17 overexpression or knockout animals (15): Specific activation of GPR17 by MDL29,951 was sufficient to arrest oligodendrocytes in an immature, nonmyelinating stage, and this effect was absent in oligodendrocyte cultures from GPR17−/− animals. From these results, we infer that MDL29,951 constitutes a powerful tool to interrogate the physiological function of orphan GPR17 despite the lack of mechanistic insight into agonist modalities such as allosterism and functional selectivity. Of equal value is pranlukast, which enhanced oligodendrocyte maturation in its own right in a strictly GPR17-dependent manner. These data suggest that the true endogenous activator of GPR17 might be present in the primary cell culture system and that pranlukast acts by counteracting this endogenous molecule. Hence, pharmacological evidence is now available to suggest that GPR17 acts as a brake to negatively regulate oligodendrocyte maturation. A major reason for remyelination failure is that OPCs do not succeed in opposing the action of intrinsic oligodendrocyte differentiation inhibitors in the lesion (2-4, 9). A role for GPR17 as intrinsic oligodendrocyte differentiation inhibitor is well in line with its associated signaling pattern because both Gαi and Gαq engagement and their downstream effector pathways may be linked to impaired oligodendrocyte maturation (59). This function of GPR17 in oligodendrocyte biology, together with the notion that its abundance is increased in active white matter plaques of MS patients and animal models of MS (15), strongly suggests that pharmacological inhibition of GPR17 signaling may be a novel strategy with high potential to promote myelin repair.
Pharmacological inhibition of GPR17 is observed already with drugs targeting the CysLT1 receptor (14, 44). However, despite the ability of CysLT1 antagonists to counteract GPR17 function in vitro, they are not suited to inclusion in a cocktail to promote remyelination in MS in vivo. This is because of both poor brain bioavailability (peripherally acting CysLT1 antagonists have not been optimized for brain penetration) and insufficient potency for GPR17. Nevertheless, our study showed that MDL29,951-triggered GPR17 activation can be completely abrogated with the CysLT1 antagonist pranlukast on both human and rodent orthologs and therefore qualify this agonist-antagonist pair at least for mechanistic studies in native or primary cells.
In conclusion, we report identification and characterization of the first synthetic small-molecule activator for the orphan GPCR GPR17. MDL29,951 is highly selective for GPR17 over the entire repertoire of receptors from the purinergic cluster and competent to stimulate GPR17 irrespective of the cellular background and expression system, in marked contrast to the activators proposed so far (14, 32). Discovery of MDL29,951 enabled us to decode GPR17 signaling along the major GPCR signaling pathways and to clarify the current controversy pertaining to its specific role in orchestrating oligodendrocyte maturation. Because GPCRs with cell type–specific expression pattern should constitute excellent targets for therapeutic intervention, GPR17 represents the first GPCR candidate for development of remyelination therapeutics to specifically relieve the remyelination block encountered in demyelinating diseases of the CNS, such as MS. In particular, we propose that future MS therapy will likely benefit from addition of GPR17 antagonists to anti-inflammatory drug cocktails that are already used in the management of MS.
MATERIALS AND METHODS
Materials and reagents
Tissue culture media and reagents were purchased from Invitrogen. CysLT1 antagonists (pranlukast and montelukast) and leukotriene D4 (LTD4) were obtained from Cayman Chemical. GV150526A was from R&D Systems, PTX was from BIOTREND Chemikalien GmbH, forskolin was from AppliChem, ASINEX compounds 1 to 5 were from ASINEX Europe BV, and the radiochemical [35S]GTPγS was from PerkinElmer Life Sciences. Restriction endonucleases and modifying enzymes were from New England Biolabs. All other laboratory reagents were obtained from Sigma-Aldrich unless otherwise specified.
Generation and origin of cDNA expression vectors
The short isoform of human GPR17 (GenBank accession no. U33447) was amplified by polymerase chain reaction (PCR) from human brain cDNA and cloned into pcDNA3.1(+)Zeo via 5′–Hind III and 3′–Xho I. The cloning strategy for the long isoform of human GPR17 (GenBank accession no. NM005291) was identical, except that it was inserted into pcDNA3.1(+). The open reading frames of rat GPR17 (GenBank accession no. DQ777767) and mouse GPR17 (GenBank accession no. AC131761) were amplified from the respective genomic DNA and inserted into pcDNA3.1(+) via 5′–Eco RI and 3′–Xho I (mGPR17) and 5′–Nhe I and 3′–Eco RV (rGPR17), respectively. For ELISA and immunofluorescence experiments, a triple HA tag (3xHA; 3xYPYDVPDYA) was introduced by PCR at the N terminus of hGPR17, and the resulting construct was inserted via 5′–Nhe I and 3′–Xho I into the pcDNA3.1(+)Zeo expression vector. To generate an hGPR17-Rluc fusion protein, the hGPR17 coding sequence without a STOP codon and Rluc were amplified, fused in frame by PCR, and subcloned into the pcDNA3.1(+)Zeo expression vector. N-terminal tagging of hGPR17 and hGPR17-Rluc with the 3xHA sequence was without effect on receptor functionality (fig. S22). GPR17-GFP2 was generated by in-frame PCR fusion of the two proteins separated by a 19–amino acid linker (GSTSPVWWNSADIQHSGGR) and inserted into pcDNA3.1(+). Fusion of GFP2 to GPR17 did not affect receptor functionality (fig. S23). Human β-arrestin2, N-terminally tagged with GFP2 (GFP2/β-arrestin2), and R. reniformis luciferase were purchased from BioSignal Packard. pcDNA3.1(+)-based expression vectors encoding the purinergic receptors P2Y1,2,4,6,11,12,13,14 and the CysLT1 receptor were from the Missouri S&T cDNA Resource Center (http://www.cdna.org/index.html). To increase surface expression of the CysLT1 receptor, its open reading frame was cloned by PCR downstream of a signal sequence FLAG-tag (MKTIIALSYIFCLVFA-DYKDDDDK) into a pcDNA3.1(+) expression vector. Gαs-188RLuc and GFP10-Gγ2 were a gift of M. Bouvier (University of Montreal, Canada). Sequence identity of all PCR-derived constructs was verified by restriction endonuclease digests and sequencing in both directions (GATC Biotech).
Commercially available and nonengineered cell lines
Human astrocytoma 1321N1 cells were purchased from the UK Health Protection Agency. Immortalized human keratinocytes (HaCat) were a gift of E. Gaffal (University of Bonn). Human blood neutrophils were isolated from human peripheral blood of healthy donors by Ficoll-Hypaque centrifugation according to standard protocols (60). All blood donors had signed an informed consent, and the study was approved by the ethics committee of the University of Bonn. Primary human keratinocytes were obtained from skin samples of healthy patients who had given an informed consent before excision. The study was approved by the local ethics committee (concession no. 090/04). All other cell lines were obtained from the American Type Culture Collection.
Cell culture and transfection of immortalized and recombinant cell lines
Native and recombinant HEK293 and 1321N1 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal calf serum (FCS), penicillin (100 U/ml), and streptomycin (100 μg/ml). For HEK-mGPR17, HEK-rGPR17, and HEK-CysLT1, the medium was supplemented with G418 (500 μg/ml) (InvivoGen), for HEK-hGPR17 and HEK-hGPR17-Rluc with zeocin (56 μg/ml) (InvivoGen), for HEK-BRET-hGPR17 with zeocin (56 μg/ml) and G418 (500 μg/ml), and for 1321N1-hGPR17 and 1321N1-hP2Y13 with G418 (800 μg/ml).
CHO-K1 cells were cultivated in DMEM: Nutrient Mixture F-12 (DMEM/F12) supplemented with 10% (v/v) FCS, penicillin (100 U/ml), and streptomycin (100 μg/ml). For Flp-In T-REx CHO cells stably expressing hGPR17 (CHO-hGPR17), medium was complemented with hygromycin B (500 μg/ml) and blasticidin (30 μg/ml) (both InvivoGen). Expression of GPR17 was induced by treatment with doxycycline (1 μg/ml) for 14 to 20 hours. For transient transfections of HEK293 cells, the Ca2+ phosphate coprecipitation method was used 24 hours after seeding. Stable cell lines were generated by Ca2+ phosphate coprecipitation or retroviral transfection essentially as described previously (61, 62) in conjunction with clonal selection using the appropriate selection antibiotics. All cells were cultivated with 5% CO2 at 37°C in a humidified atmosphere.
Cell culture of primary rat oligodendrocytes and astrocytes
Primary rat OPCs were generated from forebrains of Wistar rat pups at P1/P2 with a differential detachment method (63). Cerebrum was mechanically dissociated with a syringe and two different hollow needles (first 1.2 × 40 and then 0.60 × 30). Clump-free cell suspension was filtered through a 70-μm cell strainer (BD Biosciences), plated on poly-d-lysine–coated 75-cm2 culture flasks in DMEM supplemented with 10% (v/v) heat-inactivated FCS, penicillin (100 U/ml), and streptomycin (0.1 mg/ml) with medium exchanged every other day. After 9 to 11 days, mixed cultures were shaken overnight (230 rpm) to detach OPCs from astrocytes and microglia. The suspension was plated onto uncoated petri dishes (Corning) for 30 to 60 min to further enrich for OPCs. OPCs were then seeded into poly-l-ornithine–coated plates and maintained in serum-free proliferation medium consisting of Neurobasal with B27 (2%), GlutaMAX (2 mM), penicillin (100 U/ml), streptomycin (0.1 mg/ml), PDGF-AA (platelet-derived growth factor AA) (20 ng/ml), and bFGF (basic fibroblast growth factor) (20 ng/ml) for 2 to 5 days (37°C, 5% CO2). Thereafter, medium was switched to growth factor–free Neurobasal medium to allow for spontaneous in vitro differentiation.
Primary rat astrocytes were obtained after the mechanical removal of OPCs and maintained in DMEM supplemented with 10% (v/v) heat-inactivated FCS, penicillin (100 U/ml), and streptomycin (0.1 mg/ml).
Cell culture of primary mouse oligodendrocytes from GPR17+/− and GPR17−/− mice
Primary mouse preoligodendrocytes (O4+) were purified by immunopanning frommouse cerebral cortices as described previously (64). Briefly, dissociated cortex cells of P6 GPR17+/− or GPR17−/− mice were sequentially panned on Ran2 and GalC panning plates to deplete the astrocytes, microglia, and mature oligodendrocytes. Finally, preoligodendrocytes were enriched on the O4 panning plate. The purified preoligodendrocytes were seeded onto poly-l-ornithine–coated glass coverslips and maintained in differentiation medium [Satomedium with T3 (20 ng/ml)] for 3 days. GPR17 agonist or solvent control was added to the medium and maintained throughout the experiment.
Label-free DMR assays
DMR was recorded on a β version of the Corning Epic biosensor or the EnSpire multimode plate reader (PerkinElmer) as described in detail previously (25, 26).
Label-free bioimpedance (CellKey) assays
Cells were seeded at a density of 10,000 cells per well into 384-well biosensor plates and grown to confluence for 18 to 24 hours (37°C, 5% CO2). Before the assay, cells were washed twice with assay buffer [Hanks’ balanced salt solution (HBSS) + 20 mM Hepes] and then allowed to equilibrate for 1 hour at 37°C. The cell plate was then transferred to the CellKey (Molecular Devices), and a baseline read was recorded for 5 min before compound solutions were added simultaneously from a separate compound source plate directly onto the cell plate. Bioimpedance changes were then monitored for at least 3600 s.
[35S]GTPγS-binding assay
[35S]GTPγS-binding experiments were conducted on membranes with previously published procedures (65, 66). Different amounts of membrane proteins (HEK: 20 μg, 1321N1: 5 μg, CHO: 5 to 30 μg) were incubated with distinct concentrations of [35S]GTPγS (HEK: 0.1 nM, 1321N1: 0.07 nM, CHO: 0.2 nM), and maximum agonist-induced [35S]GTPγS loading was measured after 1 hour.
Ca2+ mobilization assays in recombinant and primary cells
Intracellular Ca2+ mobilization was quantified with the Calcium 5 Assay Kit (Molecular Devices) and the FlexStation 3 Benchtop Multimode Plate Reader. In brief, cells were seeded at a density of 50,000 cells per well (CHO and 1321N1), 60,000 cells per well (HEK), 35,000 cells per well (oligodendrocytes), or 65,000 cells per well (astrocytes) into black 96-well tissue culture plates with clear bottom (poly-d-lysine–coated for HEK cells, poly-l-ornithine–coated for oligodendrocytes and astrocytes). Cells were loaded with the Calcium 5 indicator dye for 25 to 60 min and processed according to the manufacturer’s instructions. For analysis of inhibition effects, antagonists were injected using the integrated liquid handling of the FlexStation and preincubated for 30 min.
Second messenger cAMP and IP1 accumulation assays in recombinant and primary cells
Changes of the intracellular second messengers cAMP and IP1 were quantified with the HTRF-cAMP dynamic kit and the HTRF-IP1 kit, respectively (Cisbio International), on a Mithras LB 940 reader (Berthold Technologies) according to the manufacturer’s instructions and as described previously in detail (67).
For kinetic resolution of cAMP production in real time and in living cells (Fig. 1L), the GloSensor cAMP Assay (Promega) was used following the manufacturer’s recommendations.
BRET assay
Functional BRET2 assays were performed on HEK293 cells cotransfected to stably express hGPR17-Rluc as energy donor and β-arrestin2–GFP2 as energy acceptor essentially as described in (27), except that agonist was preincubated for 5 min before addition of the Rluc substrate. Real-time analyses (0 to 140 s) of GPR17-Rluc and GFP2–β-arrestin2 interactions were performed as described in (68). Direct interaction between GPR17 and the Gαs subunit was conceived with a GPR17-GFP2 fusion protein in conjunction with Gαs-188RLuc using HEK293 cells as expression host. G protein subunit rearrangement was quantified in HEK293 cells transiently transfected to express GPR17, Gαs-188RLuc, Gγ2-GFP10 along with unlabeled Gβ1. Receptor G protein BRET and G protein activation was detected 1 min after agonist exposure.
ERK1/2 phosphorylation assay
Quantification of phosphorylated ERK1/2 levels was performed with the HTRF-Cellul’erk kit (Cisbio) following the manufacturer’s instructions, exactly as described previously (27).
ELISA quantification of tagged cell surface receptors in recombinant cells
HEK293-3xHA-GPR17 or HEK-GPR17-Rluc cells were seeded at a density of 50,000 cells per well onto poly-d-lysine–coated black 96-well plates 24 hours before analyses. Cells were fixed, permeabilized if needed, and then incubated with mouse anti-HA primary antibody (Roche; 1:400) followed by horseradish peroxidase–conjugated goat anti-mouse IgG secondary antibody (Sigma; 1:1000). Colorimetric readings were obtained using the horseradish peroxidase substrate 3,3-,5,5-tetramethylbenzidine on the Sunrise microplate absorbance reader at 450 nm. For internalization assays, cells were exposed to 10 μM agonist or its solvent for different time periods, and the amounts of internalized receptors were quantified.
Immunocytochemical detection of GPR17 in recombinant cells and primary rat oligodendrocytes and astrocytes
Cells were cultured on coated 13-mm coverslips (poly-d-lysine for HEK293, HEK-3xHA-hGPR17, and HEK-3xHA-hGPR17-Rluc, and poly-l-ornithine for OPCs and astrocytes) in 24-well dishes, fixed with 4% paraformaldehyde, permeabilized with Triton X-100 (0.1%), and blocked with 10% goat serum and 1% bovine serum albumin in phosphate-buffered saline before immunostaining with mouse anti-HA (Roche; 2 μg/ml) or rabbit anti-GPR17 (Cayman; 0.5 μg/ml) antibody, followed by Alexa Fluor 546 goat anti-mouse IgG (Sigma; 1 μg/ml) or Cy3-conjugated goat anti-rabbit (Millipore; 1 μg/ml). After mounting cells on glass slides with Mowiol, GPR17 was visualized by fluorescence microscopy with a 40× objective (Leica DM IL LED Fluo).
Immunophenotyping of primary rat oligodendrocyte cultures with specific differentiation markers
After different days in culture, primary oligodendrocytes were fixed with 4% paraformaldehyde and stained for expression of the following specific oligodendrocyte differentiation markers: NG2 for oligodendrocyte precursors, O4 for immature and mature oligodendrocytes, and MBP for mature oligodendrocytes using mouse anti-NG2 (Millipore; 1:500), mouse anti-O4 (Millipore; 1:2000), and mouse anti-MBP (Millipore; 1: 2000) as primary antibodies. Cells were then incubated for 60 min with Cy2-conjugated goat anti-mouse IgG (Millipore; 1:500). After mounting cells on glass slides with Mowiol, immunofluorescence was analyzed with fluorescence microscopy using a 40× objective (Leica DM IL LED Fluo).
Immunocytochemistry of primary mouse oligodendrocyte cultures
Differentiated oligodendrocytes from GPR17+/− and GPR17−/− mice, treated with GPR17 agonist or solvent control, were fixed for 10 min in 4% paraformaldehyde and immunostained as described previously (15). Cells were labeled with anti-MBP antibody (Santa Cruz Biotechnology) and imaged by a Zeiss LSM 510 confocal microscope. Image analysis was performed with ImageJ 1.46r.
Cerebellar slice cultures
Cerebellar slice cultures were prepared as described (69, 70). Briefly, parasagittal slices of P4 wild-type mouse cerebellum were cut at 400 mm. The slices were placed on 30-mm Millipore culture inserts with a 0.4-mm pore size in six-well plates and maintained in 50% basal medium with Earle’s salts,25%HBSS,25%horse serum, glucose (5mg/ml), and 1mMl-glutamine in the absence or presence of 30 μM MDL29,951 for 3 days.
Isolation and purification of FR900359
The selective Gαq inhibitor FR900359 was isolated from leaves of the evergreen plant Ardisia crenata with a previously published protocol (30). Structure of the Gαq inhibitor was confirmed with spectroscopic methods [nuclear magnetic resonance (NMR) and mass spectrometry; figs. S24 and S25]. Purity of the isolated compound was greater than 95% as determined by NMR and high-performance liquid chromatography. For experiments in the presence of FR900359, cells were pretreated with 1 μM final concentration for 1 hour.
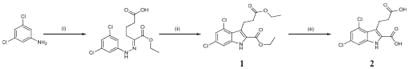
Synthesis of GPR17 agonist MDL29,951
MDL29,951 (2-carboxy-4,6-dichloro-1H-indole-3-propionic acid; product 2 in the scheme below) was synthesized by condensation of 3,5-dichloroaniline and ethyl 2-oxocyclopentanecarboxylate under Japp-Klingemann reaction conditions, followed by cyclization of the intermediate hydrazone and esterification of the carboxylate under acidic conditions, yielding intermediate 1 as previously described (71). Final saponification was performed with lithium hydroxide monohydrate in aqueous tetrahydrofuran (THF). Recrystallization from ethyl acetate/hexane gave 2 in good yield. (i) Two steps, first: NaNO2, concentrated HCl, H2O, 0°C, 15 min; second: ethyl 2-oxocyclopentanecarboxylate, NaOAc, 0°C, room temperature, 1 hour; (ii) H2SO4, EtOH, Δ, 17 hours; (iii) LiOH, THF, H2O, 17 hours.
Data analysis
Nonlinear regression and Schild analyses were performed with Prism 5.04 software (GraphPad). All label-free DMR and bioimpedance recordings are buffer- or solvent-corrected. Quantification of DMR signals was performed by calculation of the area under the curve (AUC) between 0 and 3600 s (Figs. 1, D and E, 2D, and 4A, and figs. S6C, S8D, S11, B and F, S12F, and S18) or by the maximum response within 1200 s (Fig. 2B, fig. S23B, and table S3) as appropriate. Bioimpedance traces were converted into concentration-effect curves with the peak response values within 1200 s (Fig. 2G), the AUC between 0 and 3600 s (Fig. 2E), or the AUC between 0 and 1200 s (Fig. 1G). Real-time Ca2+ recordings were transformed into concentration-effect curves with the peak response within 60 s (Figs. 1O, 2, N and P, and 6, B and D).
Statistical analyses
Comparison between two experimental groups was performed with a two-tailed Student’s t test (Fig. 7, B, C, and G) or a two-way analysis of variance (ANOVA) with Bonferroni multiple comparison test (fig. S18). P value significance thresholds were *P< 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Fig. S13. Pranlukast and montelukast do not activate rat and mouse GPR17.
Fig. S1. Sequence alignment of hGPR17 with phylogenetically related P2Y, CysLT, and small carboxylic acid receptors.
Fig. S10. Mouse and rat orthologs of GPR17 are targeted to the plasma membrane in HEK293 cells engineered to stably express these receptors.
Fig. S11. MDL29,951 productively interacts with rodent GPR17 orthologs.
Fig. S12. MDL29,951, but not ASINEX ligands 1 to 5, display functional activity on rat GPR17.
Fig. S14. Pranlukast and montelukast do not dampen Ca2+ signaling of muscarinic M3 receptors.
Fig. S15. MDL29,951 does not stimulate loading of [35S]GTPγS to membranes expressing biogenic amine histamine H3 and dopamine D2 receptors.
Fig. S16. MDL29,951 does not trigger cell activation across 16 distinct ATP-responsive immortalized and primary cell lines in a receptor panning approach.
Fig. S17. HEK293 cells express functional P2Y receptors as evidenced by their responsiveness to purinergic agonists.
Fig. S18. Forced expression of a cocktail containing eight purinergic receptors enhances functional DMR responses to purinergic agonists in HEK293 cells.
Fig. S19. Immunophenotypes of oligodendroglial cells during differentiation.
Fig. S2. GPR17 is properly trafficked to the cell surface.
Fig. S20. Pranlukast abolishes GPR17-mediated Ca2+ mobilization but does not blunt Ca2+ signaling induced by muscarinic M3 receptors in primary rat oligodendrocytes.
Fig. S21. Gαi inhibitor PTX and Gαq inhibitor FR900359 do not affect viability of primary rat oligodendrocytes.
Fig. S22. Introduction of a triple HA tag at the N termini of hGPR17 and hGPR17-Rluc does not alter signaling capacity of the receptor constructs.
Fig. S23. In-frame fusion of GFP2 to the C terminus of GPR17 does not affect receptor functionality.
Fig. S24. 1H-NMR spectrum of FR900359 in CDCl3.
Fig. S25. 13C-NMR spectrum of FR900359 in CDCl3.
Fig. S3. Native HEK293 cells are viable and respond to stimuli of endogenously expressed receptors but not to MDL29,951.
Fig. S4. Uracil nucleotides and CysLTs do not activate the long isoform of hGPR17 but promote robust activation of their established receptor targets.
Fig. S5. Pharmacological silencing of Gαq proteins attenuates GPR17-mediated cAMP formation.
Fig. S6 HEK-hGPR17 cells are unresponsive to ASINEX ligands 1 to 5 in holistic label-free DMR and Ca2+ mobilization assays.
Fig. S7. Pharmacological silencing of Gαq proteins does not abolish the capacity of MDL29,951 for dual modulation of adenylyl cyclase.
Fig. S8. GPR17-Rluc fusion protein is targeted to the cell surface and signals via G protein–mediated pathways.
Fig. S9. CysLT1 antagonists effectively inhibit intracellular Ca2+ mobilization by the CysLT1 receptor.
Table S1. Chemical structures of test compounds selected for biomolecular screening atGPR17.
Table S2. Potencies (pEC50 values) of MDL29,951 at recombinant hGPR17 cell lines.
Table S3. Functional activity of GPR17 agonist MDL29,951 on purinergic and CysLT1 receptors.
Acknowledgments
We thank I. Loef, U. Rick, and M. Vasmer-Ehses for excellent technical assistance; P. Hillmann for initial guidance of A.S. in receptor expression and calcium mobilization studies in 1321N1 cells; and Corning Inc. and PerkinElmer for their support on the Epic and EnSpire DMR readers.
Funding: This work was supported by funding from the Dr. Hilmer Foundation to S.H., a grant from the German Federal Ministry of Education and Research (BMBF) within the BioPharma Initiative “Neuroalliance” (grant no. 1615609B) to E.K., and grants from the U.S. NIH (R01NS072427 and R01NS075243) and the U.S. Army Department of Defense (W81XWH-10-1-0723) to Q.R.L.
Footnotes
Author contributions: S.H. designed, performed, and analyzed most of the functional and cell biology experiments in recombinant cells and the entire set of BRET studies, and established primary rat cell culture. L.P. designed, performed, and analyzed the biomolecular screening, label-free functional DMR, and a number of second messenger assays. A.S. designed, analyzed, and performed selected second messenger assays in 1321N1 cells and contributed to the biomolecular screening. K.S. designed, performed, and analyzed all ERK assays. R. Schröder designed, performed, and analyzed all cellular label-free bioimpedance measurements. R. Schrage and C.V. designed, performed, and analyzed all GTPγS-binding data. S.B. and N.M. designed, performed, and analyzed functional assays in primary rat oligodendrocytes and astrocytes. S.H., L.P., and N.M. cloned all human and rodent GPR17 and CysLT1 cDNA constructs and generated all stable recombinant cell lines. R.A. synthesized MDL29,951. S.K. and G.M.K. isolated the Gαq inhibitor FR900359 from natural sources. Q.R.L. and H.W. designed, performed, and analyzed all immunohistochemistry on primary oligodendrocytes from heterozygous and GPR17−/− mice. M.G. provided important ideas and assisted in project coordination. D.S., K.Z., C.D., A.P., K.M., C.E.M., and all remaining authors edited the manuscript and contributed to the discussion. J.G. and E.K. designed the research, coordinated the project, and, with contributions from all authors, wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: There are patents on the GPR17 agonist and screening assay (EP000002567698A1, US8404436B1, and US20130059899A1).
REFERENCES AND NOTES
- 1.Siffrin V, Vogt J, Radbruch H, Nitsch R, Zipp F. Multiple sclerosis—Candidate mechanisms underlying CNS atrophy. Trends Neurosci. 2010;33:202–210. doi: 10.1016/j.tins.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Franklin RJM, Ffrench-Constant C. Remyelination in the CNS: From biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- 3.Fancy SPJ, Chan JR, Baranzini SE, Franklin RJM, Rowitch DH. Myelin regeneration: A recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- 4.Aktas O, Kieseier B, Hartung H-P. Neuroprotection, regeneration and immunomodulation: Broadening the therapeutic repertoire in multiple sclerosis. Trends Neurosci. 2010;33:140–152. doi: 10.1016/j.tins.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Stys PK, Zamponi GW, van Minnen J, Geurts JJG. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. 2012;13:507–514. doi: 10.1038/nrn3275. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Diego RS, Weiner HL. Novel therapeutic strategies for multiple sclerosis— A multifaceted adversary. Nat Rev Drug Discov. 2008;7:909–925. doi: 10.1038/nrd2358. [DOI] [PubMed] [Google Scholar]
- 7.Hartung H-P, Aktas O. Evolution of multiple sclerosis treatment: Next generation therapies meet next generation efficacy criteria. Lancet Neurol. 2011;10:293–295. doi: 10.1016/S1474-4422(11)70043-6. [DOI] [PubMed] [Google Scholar]
- 8.Kremer D, Aktas O, Hartung H-P, Küry P. The complex world of oligodendroglial differentiation inhibitors. Ann Neurol. 2011;69:602–618. doi: 10.1002/ana.22415. [DOI] [PubMed] [Google Scholar]
- 9.Kuhlmann T, Miron V, Cui Q, Cuo Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- 10.Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 11.Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu T-H, Guo J, Zhang W, So K-F, Pepinsky B, Shao Z, Graff C, Garber E, Jung V, Wu EX, Wu W. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- 12.Rask-Andersen M, Almén MS, Schiöth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 13.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 14.Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, Guerrini U, Belcredito S, Cimino M, Sironi L, Tremoli E, Rovati GE, Martini C, Abbracchio MP. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006;25:4615–4627. doi: 10.1038/sj.emboj.7601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, Trapp BD, Karandikar NJ, Hsieh J, Lu QR. The oligodendrocytespecific G protein–coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat Neurosci. 2009;12:1398–1406. doi: 10.1038/nn.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, Villa G, Verderio C, Grumelli C, Guerrini U, Tremoli E, Rosa P, Cuboni S, Martini C, Buffo A, Cimino M, Abbracchio MP. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS One. 2008;3:e3579. doi: 10.1371/journal.pone.0003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fumagalli M, Daniele S, Lecca D, Lee PR, Parravicini C, Fields RD, Rosa P, Antonucci F, Verderio C, Trincavelli ML, Bramanti P, Martini C, Abbracchio MP. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J Biol Chem. 2011;286:10593–10604. doi: 10.1074/jbc.M110.162867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ceruti S, Viganò F, Boda E, Ferrario S, Magni G, Boccazzi M, Rosa P, Buffo A, Abbracchio MP. Expression of the new P2Y-like receptor GPR17 during oligodendrocyte precursor cell maturation regulates sensitivity to ATP-induced death. Glia. 2011;59:363–378. doi: 10.1002/glia.21107. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen JA, Maric D, Lau P, Barker JL, Hudson LD. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J Neurosci. 2006;26:9881–9891. doi: 10.1523/JNEUROSCI.2246-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heise CE, O’Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, Stocco R, Bellefeuille JN, Abramovitz M, Cheng R, Williams DL, Zeng Z, Liu Q, Ma L, Clements MK, Coulombe N, Liu Y, Austin CP, George SR, O’Neill GP, Metters KM, Lynch KR, Evans JF. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem. 2000;275:30531–30536. doi: 10.1074/jbc.M003490200. [DOI] [PubMed] [Google Scholar]
- 21.Maekawa A, Balestrieri B, Austen KF, Kanaoka Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc Natl Acad Sci U S A. 2009;106:11685–11690. doi: 10.1073/pnas.0905364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wunder F, Tinel H, Kast R, Geerts A, Becker EM, Kolkhof P, Hütter J, Ergüden J, Härter M. Pharmacological characterization of the first potent and selective antagonist at the cysteinyl leukotriene 2 (CysLT2) receptor. Br J Pharmacol. 2010;160:399–409. doi: 10.1111/j.1476-5381.2010.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benned-Jensen T, Rosenkilde MM. Distinct expression and ligand-binding profiles of two constitutively active GPR17 splice variants. Br J Pharmacol. 2010;159:1092–1105. doi: 10.1111/j.1476-5381.2009.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi AD, Harden TK, Nicholas RA. Is GPR17 a P2Y/leukotriene receptor? Examination of uracil nucleotides, nucleotide sugars, and cysteinyl leukotrienes as agonists of GPR17. J Pharmacol Exp Ther. 2013;347:38–46. doi: 10.1124/jpet.113.207647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schröder R, Janssen N, Schmidt J, Kebig A, Merten N, Hennen S, Müller A, Blättermann S, Mohr-Andrä M, Zahn S, Wenzel J, Smith NJ, Gomeza J, Drewke C, Milligan G, Mohr K, Kostenis E. Deconvolution of complex G protein–coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotechnol. 2010;28:943–949. doi: 10.1038/nbt.1671. [DOI] [PubMed] [Google Scholar]
- 26.Schröder R, Schmidt J, Blättermann S, Peters L, Janssen N, Grundmann M, Seemann W, Kaufel D, Merten N, Drewke C, Gomeza J, Milligan G, Mohr K, Kostenis E. Applying label-free dynamic mass redistribution technology to frame signaling of G protein–coupled receptors noninvasively in living cells. Nat Protoc. 2011;6:1748–1760. doi: 10.1038/nprot.2011.386. [DOI] [PubMed] [Google Scholar]
- 27.Bock A, Merten N, Schrage R, Dallanoce C, Bätz J, Klöckner J, Schmitz J, Matera C, Simon K, Kebig A, Peters L, Müller A, Schrobang-Ley J, Tränkle C, Hoffmann C, de Amici M, Holzgrabe U, Kostenis E, Mohr K. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat Commun. 2012;3:1044. doi: 10.1038/ncomms2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verdonk E, Johnson K, McGuinness R, Leung G, Chen Y-W, Tang HR, Michelotti JM, Liu VF. Cellular dielectric spectroscopy: A label-free comprehensive platform for functional evaluation of endogenous receptors. Assay Drug Dev Technol. 2006;4:609–619. doi: 10.1089/adt.2006.4.609. [DOI] [PubMed] [Google Scholar]
- 29.Terrin A, Di Benedetto G, Pertegato V, Cheung Y-F, Baillie G, Lynch MJ, Elvassore N, Prinz A, Herberg FW, Houslay MD, Zaccolo M. PGE1 stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: Role of compartmentalized phosphodiesterases. J Cell Biol. 2006;175:441–451. doi: 10.1083/jcb.200605050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujioka M, Koda S, Morimoto Y, Biemann K. Structure of FR900359, a cyclic depsipeptide from Ardisia crenata sims. J Org Chem. 1988;53:2820–2825. [Google Scholar]
- 31.Nesterov A, Hong M, Hertel C, Jiao P, Brownell L, Cannon E. Screening a plant extract library for inhibitors of cholecystokinin receptor CCK1 pathways. J Biomol Screen. 2010;15:518–527. doi: 10.1177/1087057110369702. [DOI] [PubMed] [Google Scholar]
- 32.Eberini I, Daniele S, Parravicini C, Sensi C, Trincavelli ML, Martini C, Abbracchio MP. In silico identification of new ligands for GPR17: A promising therapeutic target for neurodegenerative diseases. J Comput Aided Mol Des. 2011;25:743–752. doi: 10.1007/s10822-011-9455-8. [DOI] [PubMed] [Google Scholar]
- 33.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenakin TP. Cellular assays as portals to seven-transmembrane receptor-based drug discovery. Nat Rev Drug Discov. 2009;8:617–626. doi: 10.1038/nrd2838. [DOI] [PubMed] [Google Scholar]
- 36.Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Piñeyro G. β-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luttrell LM, Gesty-Palmer D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of β-blockers at the human β2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- 39.Rakesh K, Yoo B, Kim I-M, Salazar N, Kim K-S, Rockman HA. β-Arrestin–biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, Bueno OF, Vlasuk GP, Kaliman P, Bodmer R, Smith LH, Ashley E, Mercola M, Brown JH, Ruiz-Lozano P. APJ acts as a dual receptor in cardiac hypertrophy. Nature. 2012;488:394–398. doi: 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 43.Gutkind JS. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE. 2000;2000:re1. doi: 10.1126/stke.2000.40.re1. [DOI] [PubMed] [Google Scholar]
- 44.Pugliese AM, Trincavelli ML, Lecca D, Coppi E, Fumagalli M, Ferrario S, Failli P, Daniele S, Martini C, Pedata F, Abbracchio MP. Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTPγS binding and electrophysiological studies in 1321N1 cells. Am J Physiol Cell Physiol. 2009;297:C1028–C1040. doi: 10.1152/ajpcell.00658.2008. [DOI] [PubMed] [Google Scholar]
- 45.Charlton SJ, Vauquelin G. Elusive equilibrium: The challenge of interpreting receptor pharmacology using calcium assays. Br J Pharmacol. 2010;161:1250–1265. doi: 10.1111/j.1476-5381.2010.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, Ford-Hutchinson AW, Caskey CT, Evans JF. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 47.Milligan G. Orthologue selectivity and ligand bias: Translating the pharmacology of GPR35. Trends Pharmacol Sci. 2011;32:317–325. doi: 10.1016/j.tips.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Strasser A, Wittmann H-J, Buschauer A, Schneider EH, Seifert R. Species-dependent activities of G-protein-coupled receptor ligands: Lessons from histamine receptor orthologs. Trends Pharmacol Sci. 2013;34:13–32. doi: 10.1016/j.tips.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Fischer W, Franke H, Gröger-Arndt H, Illes P. Evidence for the existence of P2Y1,2,4 receptor subtypes in HEK-293 cells: Reactivation of P2Y1 receptors after repetitive agonist application. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:466–472. doi: 10.1007/s00210-005-1070-6. [DOI] [PubMed] [Google Scholar]
- 50.Wirkner K, Schweigel J, Gerevich Z, Franke H, Allgaier C, Barsoumian EL, Draheim H, Illes P. Adenine nucleotides inhibit recombinant N-type calcium channels via G protein-coupled mechanisms in HEK 293 cells; involvement of the P2Y13 receptor-type. Br J Pharmacol. 2004;141:141–151. doi: 10.1038/sj.bjp.0705588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G proteincoupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coppi E, Maraula G, Fumagalli M, Failli P, Cellai L, Bonfanti E, Mazzoni L, Coppini R, Abbracchio MP, Pedata F, Pugliese AM. UDP-glucose enhances outward K+ currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia. 2013;61:1155–1171. doi: 10.1002/glia.22506. [DOI] [PubMed] [Google Scholar]
- 53.Tucek S, Michal P, Vlachová V. Modelling the consequences of receptor–G-protein promiscuity. Trends Pharmacol Sci. 2002;23:171–176. doi: 10.1016/s0165-6147(00)01996-9. [DOI] [PubMed] [Google Scholar]
- 54.Bläsius R, Weber RG, Lichter P, Ogilvie A. A novel orphan G protein-coupled receptor primarily expressed in the brain is localized on human chromosomal band 2q21. J Neurochem. 1998;70:1357–1365. doi: 10.1046/j.1471-4159.1998.70041357.x. [DOI] [PubMed] [Google Scholar]
- 55.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davenport AP, Alexander SPH, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, Liew WC, Mpamhanga CP, Bonner TI, Neubig RR, Pin JP, Spedding M, Harmar AJ. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: Recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013;65:967–986. doi: 10.1124/pr.112.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parravicini C, Ranghino G, Abbracchio MP, Fantucci P. GPR17: Molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors. BMC Bioinformatics. 2008;9:263. doi: 10.1186/1471-2105-9-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daniele S, Trincavelli ML, Gabelloni P, Lecca D, Rosa P, Abbracchio MP, Martini C. Agonist-induced desensitization/resensitization of human G protein-coupled receptor 17: A functional cross-talk between purinergic and cysteinyl-leukotriene ligands. J Pharmacol Exp Ther. 2011;338:559–567. doi: 10.1124/jpet.110.178715. [DOI] [PubMed] [Google Scholar]
- 59.Liang X, Draghi NA, Resh MD. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J Neurosci. 2004;24:7140–7149. doi: 10.1523/JNEUROSCI.5319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sturm GJ, Schuligoi R, Sturm EM, Royer JF, Lang-Loidolt D, Stammberger H, Amann R, Peskar BA, Heinemann A. 5-Oxo-6,8,11,14-eicosatetraenoic acid is a potent chemoattractant for human basophils. J Allergy Clin Immunol. 2005;116:1014–1019. doi: 10.1016/j.jaci.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 61.Kostenis E, Martini L, Ellis J, Waldhoer M, Heydorn A, Rosenkilde MM, Norregaard PK, Jorgensen R, Whistler JL, Milligan G. A highly conserved glycine within linker I and the extreme C terminus of G protein α subunits interact cooperatively in switching G protein-coupled receptor-to-effector specificity. J Pharmacol Exp Ther. 2005;313:78–87. doi: 10.1124/jpet.104.080424. [DOI] [PubMed] [Google Scholar]
- 62.Hillmann P, Ko G-Y, Spinrath A, Raulf A, von Kügelgen I, Wolff SC, Nicholas RA, Kostenis E, Höltje H-D, Müller CE. Key determinants of nucleotide-activated G protein-coupled P2Y2 receptor function revealed by chemical and pharmacological experiments, mutagenesis and homology modeling. J Med Chem. 2009;52:2762–2775. doi: 10.1021/jm801442p. [DOI] [PubMed] [Google Scholar]
- 63.Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- 64.Chan JR, Watkins TA, Cosgaya JM, Zhang C, Chen L, Reichardt LF, Shooter EM, Barres BA. NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron. 2004;43:183–191. doi: 10.1016/j.neuron.2004.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marteau F, Le Poul E, Communi D, Communi D, Labouret C, Savi P, Boeynaems J-M, Gonzalez NS. Pharmacological characterization of the human P2Y13 receptor. Mol Pharmacol. 2003;64:104–112. doi: 10.1124/mol.64.1.104. [DOI] [PubMed] [Google Scholar]
- 66.Célanire S, Wijtmans M, Christophe B, Collart P, de Esch I, Dassesse D, Delaunoy C, Denonne F, Durieu V, Gelens E, Gillard M, Lallemand B, Lamberty Y, Lebon F, Nicolas J-M, Quéré L, Snip E, Vanbellinghen A, van Houtvin N, Verbois V, Timmerman H, Talaga P, Leurs R, Provins L. Discovery of a new class of non-imidazole oxazoline-based histamine H3 receptor (H3R) inverse agonists. ChemMedChem. 2009;4:1063–1068. doi: 10.1002/cmdc.200900055. [DOI] [PubMed] [Google Scholar]
- 67.Schröder R, Merten N, Mathiesen JM, Martini L, Kruljac-Letunic A, Krop F, Blaukat A, Fang Y, Tran E, Ulven T, Drewke C, Whistler J, Pardo L, Gomeza J, Kostenis E. The C-terminal tail of CRTH2 is a key molecular determinant that constrains Gαi and downstream signaling cascade activation. J Biol Chem. 2009;284:1324–1336. doi: 10.1074/jbc.M806867200. [DOI] [PubMed] [Google Scholar]
- 68.Galés C, Rebois RV, Hogue M, Trieu P, Breit A, Hébert TE, Bouvier M. Realtime monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- 69.Fogal B, McClaskey C, Yan S, Yan H, Rivkees SA. Diazoxide promotes oligodendrocyte precursor cell proliferation and myelination. PLoS One. 2010;5:e10906. doi: 10.1371/journal.pone.0010906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Birgbauer E, Rao TS, Webb M. Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J Neurosci Res. 2004;78:157–166. doi: 10.1002/jnr.20248. [DOI] [PubMed] [Google Scholar]
- 71.Salituro FG, Harrison BL, Baron BM, Nyce PL, Stewart KT, Kehne JH, White HS, McDonald IA. 3-(2-Carboxyindol-3-yl)propionic acid-based antagonists of the N-methyl-d-aspartic acid receptor associated glycine binding site. J Med Chem. 1992;35:1791–1799. doi: 10.1021/jm00088a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S13. Pranlukast and montelukast do not activate rat and mouse GPR17.
Fig. S1. Sequence alignment of hGPR17 with phylogenetically related P2Y, CysLT, and small carboxylic acid receptors.
Fig. S10. Mouse and rat orthologs of GPR17 are targeted to the plasma membrane in HEK293 cells engineered to stably express these receptors.
Fig. S11. MDL29,951 productively interacts with rodent GPR17 orthologs.
Fig. S12. MDL29,951, but not ASINEX ligands 1 to 5, display functional activity on rat GPR17.
Fig. S14. Pranlukast and montelukast do not dampen Ca2+ signaling of muscarinic M3 receptors.
Fig. S15. MDL29,951 does not stimulate loading of [35S]GTPγS to membranes expressing biogenic amine histamine H3 and dopamine D2 receptors.
Fig. S16. MDL29,951 does not trigger cell activation across 16 distinct ATP-responsive immortalized and primary cell lines in a receptor panning approach.
Fig. S17. HEK293 cells express functional P2Y receptors as evidenced by their responsiveness to purinergic agonists.
Fig. S18. Forced expression of a cocktail containing eight purinergic receptors enhances functional DMR responses to purinergic agonists in HEK293 cells.
Fig. S19. Immunophenotypes of oligodendroglial cells during differentiation.
Fig. S2. GPR17 is properly trafficked to the cell surface.
Fig. S20. Pranlukast abolishes GPR17-mediated Ca2+ mobilization but does not blunt Ca2+ signaling induced by muscarinic M3 receptors in primary rat oligodendrocytes.
Fig. S21. Gαi inhibitor PTX and Gαq inhibitor FR900359 do not affect viability of primary rat oligodendrocytes.
Fig. S22. Introduction of a triple HA tag at the N termini of hGPR17 and hGPR17-Rluc does not alter signaling capacity of the receptor constructs.
Fig. S23. In-frame fusion of GFP2 to the C terminus of GPR17 does not affect receptor functionality.
Fig. S24. 1H-NMR spectrum of FR900359 in CDCl3.
Fig. S25. 13C-NMR spectrum of FR900359 in CDCl3.
Fig. S3. Native HEK293 cells are viable and respond to stimuli of endogenously expressed receptors but not to MDL29,951.
Fig. S4. Uracil nucleotides and CysLTs do not activate the long isoform of hGPR17 but promote robust activation of their established receptor targets.
Fig. S5. Pharmacological silencing of Gαq proteins attenuates GPR17-mediated cAMP formation.
Fig. S6 HEK-hGPR17 cells are unresponsive to ASINEX ligands 1 to 5 in holistic label-free DMR and Ca2+ mobilization assays.
Fig. S7. Pharmacological silencing of Gαq proteins does not abolish the capacity of MDL29,951 for dual modulation of adenylyl cyclase.
Fig. S8. GPR17-Rluc fusion protein is targeted to the cell surface and signals via G protein–mediated pathways.
Fig. S9. CysLT1 antagonists effectively inhibit intracellular Ca2+ mobilization by the CysLT1 receptor.
Table S1. Chemical structures of test compounds selected for biomolecular screening atGPR17.
Table S2. Potencies (pEC50 values) of MDL29,951 at recombinant hGPR17 cell lines.
Table S3. Functional activity of GPR17 agonist MDL29,951 on purinergic and CysLT1 receptors.