

Abstract
Genes encoding the glycolytic enzymes of the facultative endocellular parasite Bartonella henselae have been analyzed phylogenetically within a very large cohort of homologues from bacteria and eukaryotes. We focus on this relative of Rickettsia prowazekii along with homologues from other α-proteobacteria to determine whether there have been systematic transfers of glycolytic genes from the presumed α-proteobacterial ancestor of the mitochondrion to the nucleus of the early eukaryote. The α-proteobacterial homologues representing the eight glycolytic enzymes studied here tend to cluster in well-supported nodes. Nevertheless, not one of these α-proteobacterial enzymes is related as a sister clade to the corresponding eukaryotic homologues. Nor is there a close phylogenetic relationship between glycolytic genes from Eucarya and any other bacterial phylum. In contrast, several of the reconstructions suggest that there may have been systematic transfer of sequences encoding glycolytic enzymes from cyanobacteria to some green plants. Otherwise, surprisingly little exchange between the bacterial and eukaryotic domains is observed. The descent of eukaryotic genes encoding enzymes of intermediary metabolism is reevaluated.
A widespread view is that genes descended from the bacterial ancestors of modern mitochondria encode housekeeping functions of modern eukaryotic cells (1, 2). According to this interpretation, sequences encoding enzymes of intermediary metabolism of the endosymbiont were transferred to the nuclear genome of the host during the evolution of mitochondria. A more specific model speculates that anaerobic syntrophy between an α-proteobacterium and a methanogenic archaean initiated the evolution of the eukaryotic cell (3). This model suggests that genes for glycolysis in modern eukaryotes descend from genes of the α-proteobacterial endosymbiont that were transferred to the nuclear genome. According to this hypothesis, the eukaryotic glycolytic enzymes should be closely related to their α-proteobacterial counterparts, as are core components of the respiratory chain and translation apparatus of mitochondrial proteomes (4–7).
The recent acquisition of the genome sequence of Bartonella henselae (C. Alsmark, C. Frank, B.C., D. Ardell, A.-S. Eriksson, A. K. Naslund, M. Holmberg, and S.G.E.A., unpublished data) provides an opportunity to study the complete glycolytic pathway of an α-proteobacterium. A close relative of Rickettsia prowazekii, B. henselae is a facultative endocellular parasite and the causative agent of cat scratch disease in humans. Like many archaea and bacteria, it lacks some of the enzymes used in the standard Embden-Meyerhof pathway. An alternative route for degrading glucose to pyruvate in prokaryotes is the Entner-Doudoroff pathway. Partial reactions of the Entner-Doudoroff pathway may interact with the Embden-Meyerhof pathway to replace steps of the standard glycolytic scheme (8). For example, the absence of phosphofructokinase in some bacteria such as B. henselae and Helicobacter pylori (9) suggests that elements of the Entner-Doudoroff pathway bypass this otherwise critical step of glycolysis. Accordingly, a complete overview of the path from glucose to pyruvate in B. henselae requires an analysis of representatives from both pathways.
We have carried out a phylogenetic analysis of 12 genes of B. henselae encoding elements in the Embden-Meyerhof and Entner-Doudoroff pathways. The results are unambiguous: none of the corresponding eukaryotic gene families clusters as a whole with their homologues among the α-proteobacteria (e.g., Agrobacteria, Bartonella, Brucella, Caulobacter, Mesorhizobium, and Zymomonas among others). Nor do any of these eukaryotic enzymes cluster as a whole with any other bacterial phylum. Indeed, there is surprisingly little evidence of horizontal gene transfer between the eukaryotic and bacterial domains among these gene families. We do observe phylogenetic anomalies that may be interpreted as examples of transfer from bacteria to eukaryotic taxa, especially the green plants, as well as from eukaryotes to some pathogenic bacteria. The present results are used to reopen a discussion of the descent of eukaryotic and bacterial coding sequences for housekeeping functions.
Materials and Methods
Informatic Analysis.
A preliminary analysis of the genome sequence data from B. henselae identified 12 genes with at least 30% amino acid identity to previously identified genes for the Embden-Meyerhof and Entner-Doudoroff pathways. Coding sequences corresponding to proteins available from the SPTR protein database (accessible as SWALL on the European Bioinformatics Institute Sequence Retrieval System (EBI SRS) server, http://srs6.ebi.ac.uk/) were collected from the European Molecular Biology Laboratory (EMBL) for each glycolytic enzyme. SPTR is composed of SWISS-PROT, SP-TrEMBL, and TrEMBL-new. In the case of glyceraldehyde-3-phosphate dehydrogenase, only sequences from SWISS-PROT were used because the number of sequences in SPTR exceeds the limit of a manageable data set. The coding sequences were then translated into the corresponding protein sequences. This process was done to avoid discrepancies between the coding sequences and amino acid sequences that in some cases are also found in the public databases. All phylogenetic trees in this report are unrooted and are computed with paup, version 4.0b8 for linux (10). Sequences were aligned with clustal-w, version 1.8 (11) with default settings.
A four-step approach was used to reduce the often large number of collected sequences: (i) The amino acid sequences were aligned. As a general rule, sequences with E-values of 10−30 or higher and 30% amino acid identity or less for the best local alignment were removed. (ii) The data set was realigned and phylogenetic trees were computed. Because the number of sequences remained high, the Neighbor Joining search algorithm was used with the Jukes and Cantor's substitution model. Five hundred bootstrap replicates were generated. (iii) Excluding some taxonomically similar representatives in nodes with high bootstrap support values (in most cases 100) further reduced the number of taxa so that only one or two taxa remain per group. For example, if a node with a bootstrap support value of 100 contains 12 representatives of green plants, only one or two of the sequences were used in the further analysis. (iv) The remaining amino acid sequences were again aligned. Regions with ambiguous alignments were manually removed with the help of the seaview software (12). The amino acids in the alignment were then replaced by the corresponding codons to produce a nucleic acid alignment. All third codon positions were excluded from the analysis.
Nucleic acid trees were calculated with a distance method using minimum evolution (ME) as criterion to minimize the sum of the branch lengths, with the maximum parsimony (MP) method and with the maximum likelihood (ML) method. Bootstrap trees were calculated with 500 replicates for the first two methods and 100 replicates for the ML method. The software modeltest was used to evaluate appropriate models and parameter values used in the ME and ML methods (13). For example, the software could help to determine whether or not to use a Γ-distribution, and if so, to estimate the relevant shape parameter, the proportion of invariable sites, and a suitable substitution model. In all searches with the ME and MP methods, random addition order was used with 10 repetitions. The topologies and the branch lengths of the nucleic acid trees were calculated with the ME method. This search was performed with 500 repetitions. The FastStep search method, which does not include branch swapping, was used in the ML bootstrap tree calculations. The limited computer processing capacity available dictated the choice of this method.
The data were also used to calculate ME and MP trees based on the amino acid sequences with the same variable regions removed as described above. The ME trees were based on the raw fraction of amino acid distances, equal rates, and no invariable sites. These searches were performed with 10 repetitions and random addition order. In both methods, 500 bootstrap replicates were generated. The topology and branch lengths in the amino acid trees correspond to a ME tree calculated with random addition order and 500 repetitions. Sequence files with accession numbers, alignments, calculations, and phylogenetic trees referred to in this report are available at our web site (which is published as supporting information on the PNAS web site, www.pnas.org) as well as at http://artedi.ebc.uu.se/Bjorn/Glycolysis/.
The Glycolytic Cohort.
The first enzyme in the standard metabolic pathway of glycolysis is hexokinase. Hexokinase catalyzes many different reactions, one of which is the conversion of glucose to glucose 6-phosphate (8). B. henselae seems not to encode hexokinase. In two other α-proteobacteria, Brucella abortus and Zymomonas mobilis, the enzyme is present; nevertheless, it is unevenly distributed in other bacterial groups as well. Among the γ-proteobacteria, hexokinase is found in Escherichia coli. Despite this, the main route for glucose in E. coli and many other bacteria is a glucose transport system that utilizes phosphoenolpyruvate as the donor to phosphorylate glucose directly to glucose-6-phosphate (8). This result may also be true for B. henselae.
Eight enzymes of the Embden-Meyerhof pathway are identifiable in B. henselae. Phylogenetic reconstructions for two of these, triose-phosphate isomerase and enolase, are presented here. The number of homologues in the public databases for phosphoglycerate mutase is insufficient for a reliable phylogenetic analysis.
The conversion of fructose 6-phosphate to fructose 1,6-bisphosphate is performed either by an ATP-dependent phosphofructokinase or in some organisms by a PPi-dependent phosphofructokinase (14). B. henselae does not encode a phosphofructokinase. An alternative route for glucose 6-phosphate in bacteria is the Entner-Doudoroff pathway (8). All four of the Entner-Doudoroff enzymes required to convert glucose-6-phosphate into glyceraldehyde-3-phosphate and pyruvate are encoded by B. henselae.
Of these four enzymes of the Entner-Doudoroff pathway, only the reconstruction for glucose-6-phosphate dehydrogenase is presented here. There are too few eukaryotic taxa for 6-phosphogluconolactonase to interpret the corresponding reconstructions. In addition, neither phosphogluconate dehydratase nor 2-dehydro-3-deoxyphosphogluconate aldolase are represented among the eukaryotic gene sequences described in the public databases.
Results
Eukaryotic Descendents of an α-Proteobacterial Ancestor.
Fig. 1 describes the nucleic acid tree of the alpha subunit of the E1 component of pyruvate dehydrogenase. All but one of the eukaryotic sequences of the subunit share a node with the seven α-proteobacterial homologues of this protein. The other bacterial homologues are separated from the α-proteobacterial-eukaryotic node by quite respectable bootstrap figures (ME, 98; MP, 100; ML, 73). This configuration confirms the identification of the eukaryotic alpha subunit of the E1 component as a descendent or sister clade of the α-proteobacterial subunit (7). The protein tree for this enzyme agrees well with Figure l.
Figure 1.

The ME tree for pyruvate dehydrogenase, E1 component, alpha subunit, based on nucleotide sequences. Taxa in brown text are eukaryotes, and taxa in blue text are bacteria. Taxa with blue background are α-proteobacteria. Taxa with green background are green plants. The bootstrap values found at nodes represent values calculated with each of three criteria: ME, parsimony, and ML, respectively. See Materials and Methods for additional details. Accession numbers, alignments, and tree files are found in supporting information.
One eukaryotic homologue, that from the algae Porphyra purpurea, is found in the bacterial domain, and it shares a node with the cyanobacterium Synechocystis sp. (Fig. 1). This anomalous but suggestive configuration is one of a small number that we observe in some of the present reconstructions. We discuss this and the other phylogenetic anomalies below.
Triose-Phosphate Isomerase.
Triose-phosphate isomerase was previously identified as the first example of a eukaryotic enzyme directly descended from α-proteobacteria (15). Although the bootstrap support is very modest in both the protein and nucleotide trees, all eukaryotic homologues of this enzyme, with the exception of Plasmodium falciparum and Trypanosoma brucei, are found in one node (Fig. 2). Five triose-phosphate isomerase orthologs from α−proteobacteria share a node. The sixth, a fragment of that enzyme from Rhizobium etli, is positioned together with a Gram-positive bacterium and a cyanobacterium. Several interpretations of this anomalous arrangement are possible (see below). However, the specific relationship between the R. etli ortholog and the eukaryotic homologues suggested by Keeling & Doolittle (15) is not discernable either in our protein tree for this enzyme or in the nucleic acid reconstruction of Fig. 2.
Figure 2.

The ME tree for triose-phosphate isomerase based on nucleotide sequences. The gray background represents an area with poorly resolved nodes. Otherwise, color coding, bootstrap values, and other details are as described in the legend to Fig. 1.
Enolase.
Archaea, Bacteria, and Eucarya are all represented in the nucleic acid (Fig. 3) and protein trees of enolase. However, the arrangement of the domains is not that seen in the ribosomal RNA trees (16). All eukaryotes are found in one well-supported node with bootstrap values of 97 and 98 (for ME and MP respectively) in the protein tree and one with values of 98, 97, and 80 in the nucleic acid tree (Fig. 3). Again, the α-proteobacteria are found in one node among other bacteria. The exact order of the divergence of the Bacteria and the Archaea cannot be resolved with the present reconstructions. We have not detected any examples of inter-domain horizontal transfer in this reconstruction nor in the protein reconstruction. In particular, there is no indication that green plants have been recipients of this enzyme from bacteria.
Figure 3.

The ME tree for enolase based on nucleotide sequences. Taxa in purple text are from Archaea. Color coding, bootstrap values, and other details are as described in the legends to Fig. 1 and Fig 2.
Glucose-6-Phosphate Dehydrogenase.
The phylogenetic reconstructions for glucose-6-phosphate dehydrogenase (G6PD) have a single node containing all eukaryotes with excellent bootstrap support in both protein (100,100) and nucleic acid (99,100,98) trees (Fig. 4). Again, all α-proteobacterial orthologs are found in one node. There are no signs of inter-domain horizontal transfers in either of the reconstructions for G6PD.
Figure 4.

The ME tree for glucose-6-phosphate dehydrogenase based on nucleotide sequences. Color coding, bootstrap values, and other details are as described in the legend to Fig. 1.
Three Supplementary Reconstructions.
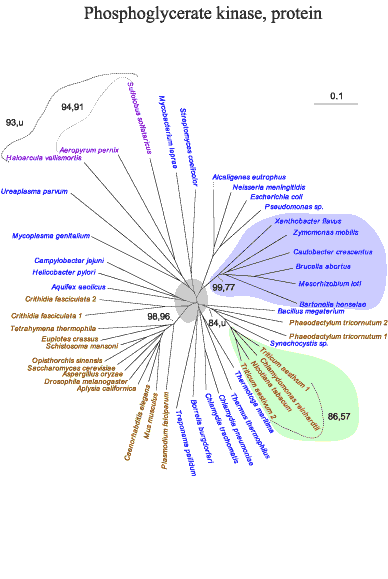
There are another five enzymes in the present cohort for which we could obtain robust protein and nucleic acid reconstructions containing both eukaryotic and bacterial clades (see Materials and Methods). Space limitations preclude presenting the corresponding reconstructions, but their characteristics are outlined below and they are presented at our web site and as supporting information. Three of these are relatively straightforward. These are the reconstructions for glyceraldehyde-3-phosphate dehydrogenase (G3PD), phosphoglycerate kinase (PGK), and pyruvate kinase (PK). For each of these, the α−proteobacteria are clustered and well separated from the eukaryotic clades. In addition, there are phylogenetic anomalies in these trees that we take up below.
There are at least two classes of G3PD (17). The reconstructions for the class I enzymes found in bacteria and eukaryotes are presented at our web site and in supporting information. The class II enzymes are found in archaea, but because their sequence identity with the class I enzymes is twenty percent or less, they have not been included in this analysis. Both the nucleic acid and the protein reconstructions for G3PD contain well-separated bacterial and eukaryotic clusters that are very strongly supported by bootstrap values in critical nodes. The αproteobacteria are found together in a well-supported node clearly separated from the eukaryotic cluster.
The protein and nucleic acid trees for PGK are characterized by poor bootstrap values for the deepest boundary between eukaryotes and prokaryotes. Nevertheless, the nodes containing eukaryotic clades other than the green plants have excellent bootstrap support in the protein tree. The archaea clades seem to be positioned among the bacteria.
The major eukaryotic node for the PK homologues is separated from that for bacteria and archaea in the protein tree as well as in the nucleotide tree. However, the bootstrap support for these nodes is poor. The α-proteobacteria share a branch well apart from the eukaryotic clades.
Paralogous Proteins.
The two remaining enzymes in our cohort provide examples of what seems to be the segregation of paralogous lineages that arose early in evolution, conceivably in the last common ancestor of eukaryotes and bacteria.
For example, inspection of the protein tree and the nucleic acid tree of glucose-6-phosphate isomerase (G6PI) suggests that both are composite trees made up of at least two paralogous lineages. Indeed, Nowitski et al. (18) have identified three putative classes of this enzyme based on sequence characteristics. Although complex, the reconstructions for G6PI do not support a special affinity between the α−proteobacterial and the eukaryotic clades.
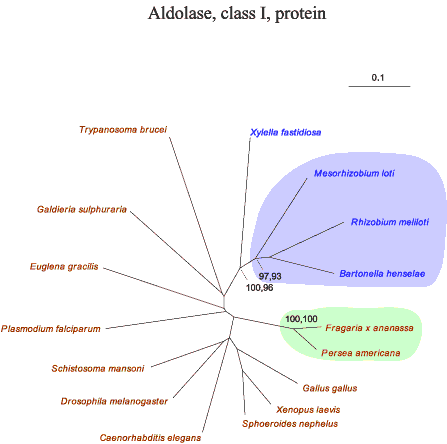
Fructose-1,6-bisphosphate aldolase (aldolase) presents the most complex reconstructions of all of the enzymes analyzed in this study. Aldolase is apparently found in three classes: Class II aldolases are found in bacteria, fungi, and some protists such as Giardia lamblia (19). Class I aldolases are found in other eukaryotes and in a limited number of bacterial taxa (17). Finally, there is a specific class I aldolase in Archaea with sequence characteristics that are clearly distinguishable from the other class I enzymes (20). Thus, ancient gene duplications, diversification, and segregation of aldolase paralogs may account for the specific distributions of these enzymes. Of course it is not excluded that other scenarios (see below) are involved as well in the phylogenetic distributions of aldolase.
The node separating bacterial and eukaryotic clades in the class I aldolase reconstruction has excellent bootstrap support values in both the protein (100,96) and nucleic acid (95,91,85) trees. Likewise, the cluster containing the class I enzymes of the α-proteobacteria is very well separated from the eukaryotic node.
In contrast, eukaryotic and bacterial taxa are intermixed in the class II trees. In the nucleic acid tree, the two types of class II aldolases are clearly distinguishable (19). The type B class contains the sequences from eukaryotes other than the fungi as well as from diverse bacteria. The type A class contains fungi and other bacteria. The node separating the types A and B have excellent bootstrap support values in the nucleic acid and protein trees. The α-proteobacteria are well represented in the type B node. Oddly, Rhizobium meliloti has one class I and one class II enzyme. Likewise, ancestral organisms may have had both class I and class II sequences, one of which was lost in their descendents to produce the modern disposition of aldolase classes.
Phylogenetic Anomalies.
There are a few anomalous clades in the reconstructions presented in Figs. 1 and 2. In particular, a few eukaryotic clades are found within the bacterial clusters. Furthermore, the reconstructions for the enzymes described at our web site and in supporting information also provide other clear examples of bacterial and eukaryotic clades that seem to be positioned in the wrong domain. Previous findings of such singularities have been interpreted as evidence for the transfer of these sequences from the endosymbiont that seeded the evolution of the mitochondria (3, 17, 18, 21, 22).
The restricted numbers of such anomalous clades speak against such an interpretation because they suggest that, if such anomalies are due to transfers, they must have been very recent ones. Indeed, some if not all of these anomalies could represent segregating paralogous sequences or phylogenetic artifacts created by variable mutation rates. We return to these below.
On the other hand, for three enzymes—G6PI, G3PD, and PGK—there is collateral evidence supporting the interpretation that bacterial sequences may have been transferred to a specific group of eukaryotes, the green plants. For each of these three enzymes, a number of plant clades are positioned within the bacterial cluster, and in association with cyanobacteria. Thus, these seemingly anomalous sequences could represent the transfer of glycolytic genes from the cyanobacterial ancestor of the chloroplasts (23) to the genomes of plant cells.
Discussion
Pairwise blast searches have been used to determine whether homologues of Archaea or Bacteria are most closely related to eukaryotic enzymes for intermediary metabolism. Typically, the bacterial homologues are found to be more closely related to the eukaryotic ones (2, 24). Gogarten et al. (1) have suggested, on the basis of such sequence comparisons, that the seeding of the mitochondrial lineages in eukaryotes was attended by a massive transfer of genes for intermediary metabolism from the ancestral endosymbiont to eukaryotes. More specifically, Martin and Müller (3) have suggested that eukaryotic glycolytic enzymes are descendents of α-proteobacterial coding sequences that were transferred to eukaryotes from an endosymbiotic ancestor of the mitochondria.
In fact, we cannot identify a single eukaryotic glycolytic enzyme family within the present cohort that clusters in a single node with α-proteobacteria or with any other modern bacterial phylum. In contrast, phylogenetic reconstructions as in Fig. 1 have identified nucleus-encoded mitochondrial enzymes that do cluster with the α-proteobacteria. Nearly 50 such mitochondrial proteins have been identified as the descendents of α-proteobacterial sequences (7, 25–27). Accordingly, the consistent absence of clustering of eukaryotic glycolytic enzymes with homologues from one or another bacterial group seems to be a significant negative result. The prior identification of the R. etli triose-phosphate isomerase as a sister clade to eukaryotic homologues (15) is not supported by our reconstructions for this gene family (see also ref. 26). In other words, the data uniformly contradict the expectations of Gogarten et al. (1) as well as of Martin and Müller (3).
Scattered findings of other individual bacterial taxa among eukaryotic clusters of enzymes have been offered as evidence for the descent of eukaryotic glycolytic enzymes from the bacterial lineage that gave rise to the mitochondria (3, 17, 18, 21, 22). In effect, these authors have interpreted the anomalous placement of a nominal bacterial homologue within a eukaryotic cluster as evidence that the bacterium is at the root of that eukaryotic lineage. If we were to consistently follow this line of reasoning, we would also be obliged to identify an isolated eukaryotic clade among bacteria as the root of the homologous bacterial cluster as in Fig. 1.
This procedure leads to flagrant contradictions. For example, the reconstructions for both G6PI and G3PD reveal bacterial clades within eukaryotic nodes and eukaryotic clades within bacterial nodes. In other words, two opposing roots should be ascribed to each of these gene families according to the reasoning of Martin and his associates (17, 18, 21, and 22). Such an arrangement might be meaningful if the phylogenetic tree were a closed circle.
We may assume for the sake of argument that all these anomalies indeed are due to gene transfer. In that case, it is far simpler to interpret nominal bacterial clades isolated in a node with eukaryotic homologues as eukaryotic coding sequences that may have been transferred to ancestors of those bacterial taxa. This parsimonious interpretation is particularly attractive because we most often observe putative transfers from eukaryotes to lineages of pathogenic bacteria. Likewise, a eukaryotic homologue isolated within a cluster of bacterial lineages could be a sequence transferred from bacteria to eukaryotic lineages. Accordingly, trees that contain both kinds of anomalies are in principle not problematic.
The reconstructions of Figs. 2–4 reveal few phylogenetic anomalies that might be interpreted as horizontal transfers between domains. Likewise, the reconstructions of the other enzymes described in supplementary information and at our web site reveal a restricted number of instances that might be interpreted as gene transfer among bacterial taxa as well as between bacteria and eukaryotes. We also observe that the α-proteobacterial enzymes tend to cluster in single nodes, which may or may not contain one or another anomalous taxon.
The most extensive putative gene transfers are seen among the green plants, and even these are limited. Putative bacterial descendents of four enzymes—namely, G6PI, G3PD, PGK, and PK—can be identified in the green plants, most often associated with cyanobacterial homologues such as those from Synechocystis. Only in the case of two of the enzymes (G6PI and PGK) are all of the “green” clades within the bacterial domain. Late transfer from a bacterial lineage after the divergence of the plants may account for the split of plant homologues between the bacterial and eukaryotic domains for the distributions of G3PD and PK.
It is often observed that phylogenetic reconstructions for genes encoding proteins tend to depart in detail from the reconstructions obtained with ribosomal RNA. Discrepancies of this sort have been offered as evidence that gene transfer is so frequent that it is meaningless to attempt the construction of genome phylogeny (28). In fact, our reconstructions seem not to be distorted by all-pervasive gene transfers. On the other hand, these phylogenetic reconstructions are definitely not isomorphic with those obtained for ribosomal RNA.
Two caveats are relevant here. First, anticipation of such discrepancies was part of the motivation for using ribosomal RNA as a universal phylogenetic marker (16). Thus, it was recognized that higher mutation rates and greater ease of transfer would make the phylogeny of proteins less robust than that of ribosomal RNA. Second, the results of random and systematic errors in tree reconstruction should not be automatically ascribed to gene transfer. In particular, gene duplications are a potential source of confusion. Thus, the divergent evolution of paralogous proteins followed by the segregation of the paralogs can generate much phylogenetic mischief. Extreme examples of the phylogenetic disorder that segregating paralogs create may be found among the different classes of aldolase and G6PI discussed above. Segregation of paralogs in related clades would be very difficult to distinguish from gene transfer or variable mutation rates.
The enzymes of the Embden-Meyerhof pathway are broadly distributed in bacteria and eukaryotes. However, archaea often exploit other enzymes for these reactions (14, 29). Homologues of a glycolytic enzyme are present in all three domains, in only three of our reconstructions (PGK, enolase, and PK). In these reconstructions, the archaeal homologues seem superficially to be phylogenetically closer to bacterial homologues than to those of the eukaryotes. However, the divergence of these domains is very difficult to resolve on the basis of current data. Notwithstanding, the putative affinities between bacterial and eukaryotic homologues have been taken as evidence for the origin of eukaryotic genes for intermediary metabolism from ancestral bacterial sequences (2, 24).
None of the trees that we have constructed for the present cohort is rooted. Nevertheless, with the exception of the enzymes found in mitochondria and chloroplasts, there is no indication that any eukaryotic gene family is rooted in modern bacterial clades, or vice versa. Indeed, all of the phylogenetic reconstructions obtained in this study are consistent with the interpretation that the divergence of the archaeal, bacterial, and eukaryotic lineages is ancient, as suggested by others (30–32). Here, “ancient” would mean that it predates the divergence of, for example, the α-proteobacteria from the other proteobacteria. If this were so, the emergence of the mitochondria would be much more recent than the divergence of eukaryotes and bacteria.
Supplementary Material
Acknowledgments
We are grateful to Manolo Gouy for hosting one of us (B.C.) in his laboratory as well as for his advice and criticism. We thank Cecilia Alsmark, Carolin Frank, Ann-Sofie Eriksson, and Kristina Näslund for sharing sequence data, and Mikael Thollesson, Carl R. Woese as well as Gary Olsen for helpful discussions. This work was supported by the Foundation for Strategic Research (to S.G.E.A.).
Abbreviations
- ME
minimum evolution
- MP
maximum parsimony
- ML
maximum likelihood
- G6PI
glucose-6-phosphate isomerase
- G3PD
glyceraldehyde-3-phosphate dehydrogenase
- PGK
phosphoglycerate kinase
- PK
pyruvate kinase
Note Added in Proof.
It has come to our attention that Huang (33) could not detect glycolysis in a strain of Bartonella quintana. This observation has no influence on the phylogenetic inferences in the present story.
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database [accession nos. AY074763–074775 (13 sequences)].
References
- 1.Gogarten J P, Olendzenski L, Hilario E, Simon C, Holsinger K E. Science. 1996;274:1750–1751. [PubMed] [Google Scholar]
- 2.Feng D F, Cho G, Doolittle R F. Proc Natl Acad Sci USA. 1997;94:13028–13033. doi: 10.1073/pnas.94.24.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin W, Müller M. Nature (London) 1998;392:37–41. doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- 4.Andersson S G E, Zomorodipour A, Andersson J O, Sicheritz-Pontén T, Alsmark U C M, Podowski R M, Näslund A K, Eriksson A-S, Winkler H H, Kurland C G. Nature (London) 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 5.Gray M W, Burger G, Lang B F. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 6.Sicheritz-Ponten T, Kurland C G, Andersson S G. Biochim Biophys Acta. 1998;1365:545–551. doi: 10.1016/s0005-2728(98)00099-1. [DOI] [PubMed] [Google Scholar]
- 7.Karlberg O, Canback B, Kurland C G, Andersson S G. Yeast. 2000;17:170–187. doi: 10.1002/1097-0061(20000930)17:3<170::AID-YEA25>3.0.CO;2-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dandekar T, Schuster S, Snel B, Huyen M, Bork P. Biochem J. 1999;343:115–124. [PMC free article] [PubMed] [Google Scholar]
- 9.Tomb J F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, et al. Nature (London) 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 10.Swofford D L. paup*. Phylogenetic Analysis Using Parsimony (*and Other Methods. Sunderland, MA: Sinauer; 1999. , Version 4. [Google Scholar]
- 11.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galtier N, Gouy M, Gautier C. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 13.Posada D, Crandall K A. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 14.Siebers B, Klenk H-P, Hensel R. J Bacteriol. 1998;180:2137–2143. doi: 10.1128/jb.180.8.2137-2143.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keeling P J, Doolittle F D. Proc Natl Acad Sci USA. 1997;94:1270–1275. doi: 10.1073/pnas.94.4.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woese C R. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin W, Schnarrenberger C. Curr Genet. 1997;32:1–18. doi: 10.1007/s002940050241. [DOI] [PubMed] [Google Scholar]
- 18.Nowitzki U, Flechner A, Kellermann J, Hasegawa M, Schnarrenberger C, Martin W. Gene. 1998;214:205–213. doi: 10.1016/s0378-1119(98)00229-7. [DOI] [PubMed] [Google Scholar]
- 19.Henze K, Morrison H G, Sogin M L, Muller M. Gene. 1998;222:163–168. doi: 10.1016/s0378-1119(98)00499-5. [DOI] [PubMed] [Google Scholar]
- 20.Siebers B, Brinkmann H, Dorr C, Tjaden B, Lilie H, van der Oost J, Verhees C H. J Biol Chem. 2001;276:28710–28718. doi: 10.1074/jbc.M103447200. [DOI] [PubMed] [Google Scholar]
- 21.Figge R M, Schubert M, Brinkmann H, Cerff R. Mol Biol Evol. 1999;16:429–440. doi: 10.1093/oxfordjournals.molbev.a026125. [DOI] [PubMed] [Google Scholar]
- 22.Henze K, Badr A, Wettern M, Cerff R, Martin W. Proc Natl Acad Sci USA. 1995;92:9122–9126. doi: 10.1073/pnas.92.20.9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray M W. Int Rev Cytol. 1992;141:233–357. doi: 10.1016/s0074-7696(08)62068-9. [DOI] [PubMed] [Google Scholar]
- 24.Doolittle R F, Feng D-F, Tsang S, Cho G, Little E. Science. 1996;271:470–477. doi: 10.1126/science.271.5248.470. [DOI] [PubMed] [Google Scholar]
- 25.Macrotte E M M, Xenarios I, van der Bliek A M, Eisenberg D. Proc Natl Acad Sci USA. 2000;97:12115–12120. doi: 10.1073/pnas.220399497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurland C G, Andersson S G. Microbiol Mol Biol Rev. 2000;64:786–820. doi: 10.1128/mmbr.64.4.786-820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray M W, Burger G, Lang B F. Genome Biol. 2001;2:1018.1–1018.5. doi: 10.1186/gb-2001-2-6-reviews1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doolittle W F. Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- 29.de Vos W M, Kengen W M, Voorhorst W G B, van der Oost J. Extremophiles. 1998;2:201–205. doi: 10.1007/s007920050061. [DOI] [PubMed] [Google Scholar]
- 30.Kyrpides N C, Olsen G J. Trends Genet. 1998;14:442–444. [Google Scholar]
- 31.Sicheritz-Ponten T, Andersson S G. Nucleic Acids Res. 2001;29:545–552. doi: 10.1093/nar/29.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forterre P. C R Acad Sci Ser III. 2001;324:1067–1076. doi: 10.1016/s0764-4469(01)01403-2. [DOI] [PubMed] [Google Scholar]
- 33.Huang K Y. J Bacteriol. 1967;93:853–859. doi: 10.1128/jb.93.3.853-859.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}